


Preamble
Inflammation is a normal defence mechanism that protects the host from infection and other insults; it initiates pathogen killing as well as tissue repair processes and helps to restore homeostasis at infected or damaged sites. It is typified by redness, swelling, heat, pain and loss of function, and involves interactions among many cell types and the production of, and responses to, a number of chemical mediators. Self-regulation of the inflammatory response involves the activation of negative feedback mechanisms such as the secretion of anti-inflammatory cytokines, inhibition of pro-inflammatory signalling cascades, shedding of receptors for inflammatory mediators and activation of regulatory cells. As such, and controlled properly, regulated inflammatory responses are essential to remain healthy and maintain homeostasis. Pathological inflammation involves a loss of tolerance and/or of regulatory processes. Where this becomes excessive, irreparable damage to host tissues and disease can occur(Reference Calder, Albers and Antoine1). Such diseases are characterised by markedly elevated concentrations of inflammatory markers and of activated inflammatory cells at the site of tissue damage and in the systemic circulation. While the existence of inflammatory diseases has been long recognised, it is only more recently that the condition of chronic low-grade inflammation has received attention, particularly in relation to obesity, the metabolic syndrome and CVD. Chronic low-grade inflammation is characterised by raised concentrations of inflammatory markers in the systemic circulation. This article sets out to explain the nature of chronic low-grade inflammation in the context of overweight and obesity, and to describe the factors that might influence it, in particular those related to diet. The literature in the areas of adipose tissue, obesity and inflammation, and dietary components and inflammation is vast, and it is not possible to mention all studies here. In particular, the review of diet and its components and inflammation is not exhaustive, although the main studies of relevance are included.
Concept and markers of low-grade inflammation
Obesity and low-grade inflammation
The concept of systemic, chronic, but low-grade inflammation as a risk factor for the metabolic syndrome and for type 2 diabetes is based on the observation of elevated blood levels of inflammation-associated markers in people with incident type 2 diabetes or with the metabolic syndrome(Reference Kolb and Mandrup-Poulsen2, Reference Fernandez-Real and Pickup3). The up-regulation of systemic indicators of inflammation such as leucocyte count, and serum and plasma concentrations of acute-phase proteins, pro-inflammatory cytokines, chemokines, soluble adhesion molecules and prothrombotic mediators is modest, usually less than 2-fold above what is observed in controls. Diagnostic criteria for low-grade inflammation have not been precisely defined, but the phenotype per se is not disputed.
Systemic concentrations of pro-inflammatory mediators are higher in obese (BMI >30 kg/m2) than in normal-weight persons(Reference Herder, Peltonen and Koenig4, Reference Herder, Illig and Rathmann5). Serum or plasma concentrations of TNF-α or IL-6 in healthy adults are typically 0·01–2 pmol/l(Reference Muller, Martin and Koenig6). Other inflammatory mediators, such as monocyte chemoattractant protein (MCP)-1, interferon (IFN)-γ-induced protein-10 and IL-18, may reach mean concentrations of 10 pmol/l; macrophage migration inhibitory factor (MIF) and regulated on activation, normal T expressed and secreted (RANTES) concentrations may get close to the nanomolar range; and C-reactive protein (CRP) concentration is often above 10 nmol/l. The variation in concentrations of most mediators among non-obese or obese individuals is at least 10-fold. Hence, there is a substantial overlap between non-obese and obese persons. However, there is a positive relationship between BMI and other measures of obesity such as waist circumference and circulating concentrations of CRP and other inflammatory markers(Reference Kim, Park and Kawada7).
A mechanistic link between obesity and low-grade inflammation was first proposed by Hotamisligil et al. (Reference Hotamisligil, Shargill and Spiegelman8) who showed that white adipose tissue synthesises and releases the pro-inflammatory cytokine TNF-α. The expression of TNF-α is elevated in adipocytes of obese and insulin-resistant mice, while insulin sensitivity is improved following administration of anti-TNF-α antibodies. Based on these data, it was suggested that adipose tissue plays an important immune role and may be a major source of pro-inflammatory mediators which initiate the development of chronic inflammation, insulin resistance and atherosclerosis, all of which are characteristics of the metabolic dysregulation associated with obesity. The discovery of leptin modified the view of adipose tissue as being an ‘inert’ energy store to being the largest endocrine gland in the body. Leptin is produced and secreted by white adipose tissue. The discovery of leptin introduced the concept of ‘adipocytokines’ or ‘adipokines’, substances produced by adipose tissue and which circulate in the bloodstream, so exerting systemic effects as hormones(Reference Trayhurn and Wood9). Some adipokines are produced within adipose tissue exclusively by adipocytes (e.g. leptin, adiponectin, serum amyloid A (SAA)), while others are produced by both adipocytes and other cell types of the non-adipocyte fraction of adipose tissue. It is now recognised that macrophages accumulate in the adipose tissue in obesity(Reference Tilg and Moschen10, Reference Wellen and Hotamisligil11) (Fig. 1) and that these may represent major contributors to the production of adipokines(Reference Koerner, Kratzsch and Kiess12, Reference Trujillo and Scherer13).
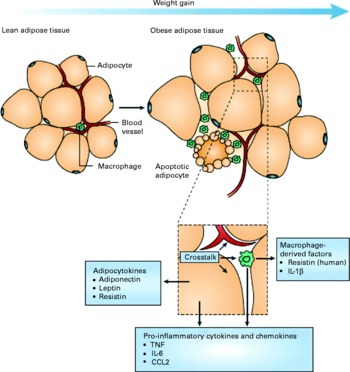
Fig. 1 Schematic representation of the interaction between adipocytes and macrophages showing some of the molecules released. Expansion of adipose tissue during weight gain leads to the recruitment of macrophages through various signals (e.g. chemokines such as chemokine (C–C motif) ligand 2 (CCL2)) released by adipocytes. Macrophages accumulate around apoptotic adipocytes. Mediators synthesised by adipocytes and resident macrophages contribute to local and systemic inflammation. Reproduced with permission from Tilg & Moschen(Reference Tilg and Moschen10).
Adipose tissue as a source of inflammatory mediators
Adipose tissue expresses and secretes into the systemic circulation a growing list of hormones, inflammatory mediators and immune system effectors. The products of adipose tissue can be categorised into several groups (Table 1):
(1) Hormones: many of the hormones produced by adipose tissue affect the immune system and insulin sensitivity. Leptin appears to be pro-inflammatory(Reference Stofkova14), while adiponectin appears to be anti-inflammatory and insulin-sensitising(Reference Gil-Campos, Canete and Gil15). Similarly, visfatin(Reference Fukuhara, Matsuda and Nishizawa16) and resistin(Reference McTernan, Kusminski and Kumar17) contribute to the development of insulin resistance, while omentin(Reference Yang, Lee and Hu18, Reference Yang, Lee and Hu19) appears to be an insulin sensitiser. It should be noted that most studies identifying the roles of these hormones have been performed in rodents and the immune and insulin-sensitising effects of these hormones in humans remain unclear(Reference Sethi and Vidal-Puig20, Reference Stephens and Vidal-Puig21).
(2) Acute-phase proteins: these proteins are secreted in the acute phase of inflammation and include plasminogen activator inhibitor 1 (PAI-1)(Reference Juhan-Vague, Alessi and Mavri22), pentraxine-3, lipocalin 24p3, haptoglobin, SAA(Reference Poitou, Viguerie and Cancello23) and α1-glycoprotein.
(3) Cytokines: these are the classic peptide mediators of inflammation and include IL-1, IL-1 receptor antagonist (IL-1ra)(Reference Somm, Cettour-Rose and Asensio24–Reference Meier, Bobbioni and Gabay26), IL-6, IL-7, IL-18(Reference Netea, Joosten and Lewis27–Reference Straczkowski, Kowalska and Nikolajuk30), IL-10(Reference Cancello, Henegar and Viguerie31, Reference Juge-Aubry, Somm and Pernin32), MIF(Reference Skurk, Herder and Kraft33) and TNF-α.
(4) Chemokines: these include IL-8(Reference Straczkowski, Dzienis-Straczkowska and Stepien34, Reference Straczkowski, Kowalska and Nikolajuk35), MCP-1, -3, -4, RANTES (now known as chemokine (C–C motif) ligand (CCL) 5), angiopoietin, metallothioneins, macrophage inflammatory protein (MIP)-1α and -1β (now known as CCL3 and CCL4, respectively)(Reference Curat, Wegner and Sengenes36), and induced protein-10(Reference Herder, Hauner and Kempf37).
(5) Growth factors: transforming growth factor (TGF)-β(Reference Fain, Tichansky and Madan38).
(6) Components of the alternative complement system: adipsin and factors C2, C3, C4, C7, B and D (these are expressed more highly in omental compared with subcutaneous adipose tissue(Reference Gabrielsson, Johansson and Lonn39, Reference Guerre-Millo40)).
(7) Retinol-binding protein 4 which is linked with insulin resistance(Reference Graham, Yang and Bluher41, Reference Yang, Graham and Mody42), although its precise role has been debated(Reference Janke, Engeli and Boschmann43).
Table 1 Cytokines expressed or secreted by human adipose tissue

MCP, monocyte chemoattractant protein; CCL, chemokine (C–C motif) ligand; RANTES, regulated on activation, normal T expressed and secreted; MIP, macrophage inflammatory protein; IL-1ra, IL-1 receptor antagonist; IP, interferon-γ-induced protein; TGF, transforming growth factor.
Adipose tissue distribution and its impact on inflammation
Obesity is characterised by an expansion of the mass of adipose tissue and dramatic changes in its distribution in the body. Simplistically speaking, accumulation of adipose tissue in the thorax and abdomen (variously termed abdominal, central, visceral, splanchnic or android obesity) results in an increased risk for diabetes and atherosclerosis, while excess adipose tissue in the lower part of the body (termed gynoid obesity) does not appear to be associated with major metabolic consequences(Reference Carey, Walters and Colditz44, Reference Kissebah, Vydelingum and Murray45). The increase in abdominal fat mass is associated with a chronic elevation of the circulating concentrations of inflammatory mediators including several acute-phase inflammatory proteins such as CRP(Reference Visser, Bouter and McQuillan46, Reference Cook, Mendall and Whincup47), pro- and anti-inflammatory cytokines, adhesion molecules and pro-thrombotic molecules(Reference Juhan-Vague, Alessi and Mavri22, Reference Cook, Mendall and Whincup47–Reference Wellen and Hotamisligil49). It should be noted that the liver and the lymphoid organs are usually the major production sites of these inflammatory mediators but in obesity, adipose tissue becomes a major producer resulting in a chronic and constant local and systemic inflammatory milieu (Table 2). Abdominal obesity is a risk factor for type 2 diabetes, hypertension, dyslipidaemia and CVD(Reference Despres and Lemieux50), and also probably obesity-associated hepatic diseases (non-alcoholic fatty liver disease and non-alcoholic steatohepatitis). Glucose intolerance is significantly more common in subjects with abdominal obesity compared with those with fat mass accumulation in their lower part of the body. Plasma TAG concentrations are also significantly more elevated in individuals with abdominal obesity. It appears that the anatomical localisation of adipose tissue is of paramount importance in relation to its physiological function, i.e. handling of lipids (lipogenesis, lipolytic activity), expression of multiple genes, and response to insulin, catecholamines, sex hormones and cortisol(Reference Lafontan and Girard51). In addition, the profile of adipokines produced is dissimilar between the subcutaneous and abdominal adipose tissues. Thus, leptin is preferentially expressed and secreted by subcutaneous adipose tissue(Reference van Harmelen, Dicker and Ryden52), while the expression of adiponectin, visfatin, omentin, resistin, PAI-1, IL-8, IL-7, IL-1α, MCP-1, TGF-β, growth-related oncogen-α, CCL5 and MIP-1β is higher in abdominal fat. In contrast to such distributions, there are reports that IL-6 and TNF-α seem to be equally synthesised by the different sites(Reference Skurk, Kolb and Muller-Scholze28, Reference Curat, Wegner and Sengenes36, Reference Alessi, Bastelica and Morange53–Reference Arner59). It is important to mention that in severe obesity, the part played by the abdominal or the very abundant subcutaneous adipose tissue in the systemic delivery of inflammatory mediators is still not well understood. Nevertheless, the distinct profile of adipokine secretion between the abdominal and subcutaneous adipose tissues probably contributes to the increased risk of metabolic and cardiovascular complications and to the development of other complications such as hepatic steatosis and non-alcoholic steatohepatitis in obese individuals. Finally, other adipose tissue depots in so-called ‘ectopic sites’, such as within the liver, heart or skeletal muscle, may contribute to the production of inflammatory mediators in the absence of obesity. In this regard, the local production of the inflammatory molecules by adipose tissue within the heart may be important; the amount of this tissue and its proximity to the coronary vessels could contribute to the development of coronary pathologies(Reference Rabkin60, Reference Silaghi, Piercecchi-Marti and Grino61).
Table 2 Modification of circulating inflammatory marker concentrations in relation to obesity and weight loss

Cell populations of adipose tissue
Adipose tissue is a heterogeneous tissue composed of several cell types: mature adipocytes, pre-adipocytes, fibroblasts, endothelial cells, mast cells, granulocytes, lymphocytes and macrophages. Cells within adipose tissue, apart from mature adipocytes, are collectively termed the stroma-vascular fraction. The various cell types have not been precisely characterised, nor has the relative change of their quantitative contributions to the tissue in obesity. Because of the heterogeneity of cells in the adipose tissue, the cellular source of the inflammatory factors secreted by the tissue into the systemic circulation remains unknown. In vitro studies have demonstrated that mature adipocytes express inflammatory factors such as TNF-α(Reference Fain, Madan and Hiler56). SAA is overexpressed and secreted in abundance by isolated adipocytes from obese subjects, as is leptin, while secretion of adiponectin is suppressed. SAA and leptin production by adipose tissue depends on adipocyte size(Reference Yang, Lee and Hu18, Reference Yang, Lee and Hu19, Reference Poitou, Viguerie and Cancello23, Reference Jernas, Palming and Sjoholm62). Adipocyte size also influences the expression of other inflammatory mediators as demonstrated by fractionation studies of adipocytes coupled with studies of gene expression profiles(Reference Jernas, Palming and Sjoholm62). Adipocyte size, for example, determines the production of IL-6, IL-8, MCP-1 and granulocyte colony-stimulating factor(Reference Skurk, berti-Huber and Herder29). Although adipocyte hypertrophy precedes the development of type 2 diabetes(Reference Weyer, Foley and Bogardus63), a growing number of studies indicate that the principal site of production of inflammatory mediators appears to be the stroma-vascular fraction(Reference Curat, Wegner and Sengenes36, Reference Fain64–Reference Xu, Barnes and Yang67). More recent studies in mice have suggested that the infiltration of obese adipose tissue by macrophages is accompanied or even preceded by an influx of T-lymphocytes(Reference Henegar, Tordjman and Achard68–Reference Wu, Ghosh and Perrard71) and that T-cells have a key early role(Reference Kintscher, Hartge and Hess69). Early work indicated the presence of CD3-positive T-lymphocytes in human adipose tissue(Reference Bornstein, Abu-Asab and Glasow72), and more recent studies have shown high numbers of T-cells in the adipose tissue of diet-induced obese insulin-resistant mice(Reference Wu, Ghosh and Perrard71). Furthermore, Wu et al. (Reference Wu, Ghosh and Perrard71) demonstrated the presence of CD3-positive T-lymphocytes in human adipose tissue and described the expression of RANTES, a T-cell-specific chemokine, and its respective receptor CCR5 in the visceral adipose tissue of morbidly obese patients. A recent study in mice reported mainly CD8-positive lymphocyte infiltration in hypoxic areas within the adipose tissue(Reference Rausch, Weisberg and Vardhana70). Most recently, it has been shown that pro-inflammatory T-lymphocytes are present in visceral adipose tissue and may contribute to local inflammatory cell activation before the appearance of macrophages, suggesting that these cells could play an important role in the initiation and perpetuation of adipose tissue inflammation as well as the development of insulin resistance(Reference Kintscher, Hartge and Hess69).
It has been proposed that macrophages and mature adipocytes are derived from the same precursor cells and show close gene expression profiles including the Toll-like receptors (TLR). Pre-adipocytes exert ‘macrophage-like’ effects when exposed to strong pro-inflammatory environments(Reference Cousin, Munoz and Andre73, Reference Cousin, Andre and Casteilla74). It should be noted, however, that the vast majority of the macrophage infiltration in adipose tissue in obesity originates most probably from the circulation. In obese subjects, these macrophages typically aggregate in crowns around apoptotic adipocytes(Reference Cancello, Henegar and Viguerie31) (Figs. 1 and 2). Although these macrophages express activation markers, they could be pro- or anti-inflammatory depending on the degree of obesity and its evolution as suggested by studies in mice showing that weight gain is accompanied by transformation from a macrophagic M2 (anti-inflammatory) phenotype towards an M1 (pro-inflammatory) profile(Reference Lumeng, Bodzin and Saltiel75) (Fig. 3)(Reference Olefsky and Glass76). Consequently, secretion profiles of the adipose tissue can change depending on the phenotype of the cell population infiltrating it during the different stages of obesity (initiation, aggravation, maintenance and weight loss; Fig. 4)(Reference Karastergiou and Mohamed-Ali77).

Fig. 2 Adipose tissue from non-obese and obese human subjects showing macrophage infiltration. Macrophages are stained with HAM56 antibody. Reproduced with permission from Cancello et al. (Reference Cancello, Tordjman and Poitou65).

Fig. 3 Schematic representation of factors regulating macrophage polarity and insulin resistance in adipose tissue. Under lean conditions, adipocytes secrete factors, such as IL-13, that promote alternative activation of macrophages. Alternatively activated (M2) macrophages secrete anti-inflammatory mediators, such as IL-10, and may secrete insulin-sensitising factors. Obesity induces changes in adipocyte metabolism and gene expression, resulting in increased lipolysis and the release of pro-inflammatory NEFA and factors that recruit and activate macrophages, such as chemokines and TNF-α. Activated M1 macrophages produce large amounts of pro-inflammatory mediators, such as TNF-α, IL-1β and resistin, that act on adipocytes to induce an insulin-resistant state. This establishes a positive feedback loop that further amplifies inflammation and insulin resistance. IFN, interferon; LPS, lipopolysaccharide. Reproduced with permission from Olefsky & Glass(Reference Olefsky and Glass76).

Fig. 4 Schematic representation of the alterations in adipose tissue that accompany body-weight gain. In the lean state, the tissue secretes elevated levels of adiponectin and other anti-inflammatory adipokines and is insulin responsive. Energy intake in excess of expenditure is followed by adipocyte hypertrophy and death and chemotactic adipokine release (see Fig. 1). This facilitates macrophage infiltration into the tissue and exacerbates the inflammatory response. These secretory changes are accompanied by local insulin resistance and hypoxia. Many of the adipokines released by inflamed adipose tissue cause insulin resistance and endothelial dysfunction. COX, cyclo-oxygenase; HGF, hepatocyte growth factor; MIF, macrophage migration inhibitory factor; MMP, matrix metalloproteinase; NGF, nerve growth factor; NOS, NO synthase; PAI-1, plasminogen activator inhibitor-1; RANTES, regulated on activation, normal T expressed and secreted; SAA, serum amyloid A; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; MCP, monocyte chemoattractant protein; RAS, renin-angiotensin system. Reproduced with permission from Karastergiou & Mohamed-Ali(Reference Karastergiou and Mohamed-Ali77).
Adipose tissue macrophages may contribute to the maintenance of the low-grade inflammatory state linked to obesity(Reference Curat, Wegner and Sengenes36). Factors that induce the infiltration and activation of macrophages in the adipose tissue are probably multifactorial. Paracrine, autocrine and endocrine signals, as well as mechanical modifications (hypertrophy and adipocyte hyperplasia), could play a role in this phenomenon. Many adipokines synthesised by the adipose tissue are candidates to attract inflammatory cells. In vitro studies have suggested that leptin itself (at supra-physiological levels) induces adhesion proteins, hence facilitating the migration of monocytes(Reference Curat, Miranville and Sengenes78). Conversely, adiponectin may inhibit this process(Reference Guzik, Mangalat and Korbut79). Very little is known about the role of selectins, integrins and elements of adhesion to the extracellular matrix, in the process of macrophage attraction to the adipose tissue. Gene expression studies with human adipose tissue have demonstrated that the levels of expression of MCP-1, colony-stimulating factor-3 and the urokinase plasminogen activator CD87 increase significantly in the adipose tissue of subjects with morbid obesity(Reference Cancello, Henegar and Viguerie31). MCP-1 is a strong chemoattractant and it acts via its receptor CCR2. MCP-1 is synthesised predominantly by macrophages and endothelial cells and, to a lesser extent, by adipocytes. In one study, CCR2 gene knockout mice showed a reduction of macrophage infiltration in the adipose tissue and improvement of insulin sensitivity(Reference Weisberg, Hunter and Huber80). This led to the suggestion that MCP-1 and its receptor CCR2 are major players in the macrophage accumulation within the adipose tissue(Reference Weisberg, Hunter and Huber80). However, contradictory data suggest that MCP-1 might not be such a crucial candidate(Reference Inouye, Shi and Howard81). The role of MCP-1 in the macrophage accumulation in human obesity needs to be established. Other candidate molecules and other mechanisms continue to be explored. Local hypoxia could also play an important role in the attraction and retention of macrophages within the adipose tissue(Reference Trayhurn, Wang and Wood82). Hypoxia-inducible factor-1α, a transcription factor normally induced by hypoxia, is overexpressed in the subcutaneous adipose tissue of obese subjects and its expression is decreased during weight reduction(Reference Cancello, Henegar and Viguerie31). Tissue hypoxia induces macrophage attraction into solid tumours as well as into atherosclerotic plaques. Adipose tissue of obese subjects could be hypoxic in some areas and a local expression of chemokines could be induced. It should be noted that leptin, which possesses indirect chemoattractant properties, is induced by hypoxia(Reference Guerre-Millo, Grosfeld and Issad83).
It is generally considered that macrophage accumulation in adipose tissue is detrimental. However, macrophage accumulation could be related to an adaptation process associated with the augmentation of fat mass, and macrophage accumulation could be necessary for the upkeep of the tissue and perhaps to limit its expansion. It appears that macrophage aggregates within the adipose tissue are localised around apoptotic adipocytes, suggesting that one of their functions is to clean up the debris of dying and dead cells(Reference Cinti, Mitchell and Barbatelli84). In addition to their role in cleaning up the old cells, the accumulation of macrophages may also be useful for the formation of new vessels, particularly at the site of inflammation and in ischaemic zones when adipose tissue grows(Reference Bouloumie, Casteilla and Lafontan85). Macrophages also control fat mass growth and modify the biology of adipocytes and pre-adipocytes in a paracrine manner. The specific effects of TNF-α and IL-6 on different adipocyte functions (increased lipolysis, modification of adipocyte secretion patterns and induction of adipocyte insulin resistance) have been shown(Reference Lagathu, Bastard and Auclair86). In the presence of a medium derived from human macrophages, human pre-adipocytes showed a drastic change in their phenotype, acquiring a pro-inflammatory phenotype and secreting significant amounts of IL-6 and IL-8, and they grew well(Reference Lacasa, Taleb and Keophiphath87), but differentiated poorly(Reference Lacasa, Taleb and Keophiphath87, Reference Constant, Gagnon and Landry88). These cellular alterations are induced in co-cultures without direct cellular contact, suggesting the key role of soluble factors, although it cannot be excluded that a direct cell–cell interaction also plays a role in modifying pre-adipocyte or adipocyte biology. The mature adipocyte also endures profound modifications of its biology in culture systems with a medium from activated macrophages. Other than the pro-inflammatory state, increased lipolysis and resistance to insulin have been observed(Reference Permana, Menge and Reaven89, Reference Suganami, Nishida and Ogawa90). TNF-α has been proposed to mediate these effects. From the molecular point of view, the NF-κB pathway, implicated in the primary regulation of inflammatory responses(Reference Kumar, Takada and Boriek91–Reference Sigal93) (Fig. 5)(Reference Maury and Brichard94), is induced in the pre-adipocyte(Reference Lacasa, Taleb and Keophiphath87) and in the adipocyte in the presence of a medium from macrophages(Reference Suganami, Tanimoto-Koyama and Nishida95). The NF-κB pathway is also brought into play in macrophages in contact with a medium from adipocytes. TLR appear to be important players which lead to the induction or suppression of genes orchestrating the inflammatory response. TLR-4 is the bacterial lipopolysaccharide (LPS) receptor, but data have shown that the NEFA produced by adipocytes after adrenergic stimulation are also strong inducers of the TLR-4/NF-κB system(Reference Suganami, Tanimoto-Koyama and Nishida95) (Fig. 5). TLR-4 is expressed by adipocytes and is overexpressed during obesity(Reference Lin, Lee and Berg96). TLR-4 knockout mice are protected from insulin resistance induced by lipid infusions(Reference Shi, Kokoeva and Inouye97).

Fig. 5 Schematic representation of the cross-talk between adipocytes and macrophages of adipose tissue in obesity. TNF-α produced by macrophages activates adipocytes via TNF-α-receptor-1 (TNFR1) and the NF-κB pathway. TNF-α also induces lipolysis leading to the release of NEFA. Saturated NEFA in turn activate the Toll-like receptor 4 (TLR4)/NF-κB pathway in both macrophages and adipocytes, thereby further amplifying the inflammatory process. Some of the adipokines produced (e.g. monocyte chemoattractant protein-1 (MCP-1)) exert chemoattractant activity through binding to specific receptors (CXC chemokine receptor (CXCR) and CC chemokine receptor (CCR)) of macrophages, leading to their infiltration in obese adipose tissue. Reproduced with permission from Maury & Brichard(Reference Maury and Brichard94).
Based on these different studies, a dual effect of macrophages of the adipose tissue could be expected: first, a local ‘beneficial’ effect in clearing out old adipocytes, and in the control and of the development of fat mass and second, a deleterious local and systemic effect via the increase in the production and secretion of adipokines, promoting the progression of complications of obesity and the induction of insulin resistance.
Adipokines and chronic low-grade inflammation
Adipokines and the complications of obesity
Inflammatory molecules are likely candidates exerting a molecular link between the adipose tissue and the metabolic, cardiovascular, hepatic and thrombotic complications, and certain cancer types occurring in conjunction with or as a consequence of human obesity. A myriad of candidate adipokines are proposed to play this role(Reference Matsuzawa98–Reference Chen100). In the cardiovascular field, they can be considered as risk factors, and even directly play a pathophysiological role favouring the initiation and progression of atherosclerosis. Relationships between abnormalities of cardiac function in obese subjects, the accumulation of abdominal fat and low-grade inflammation have been suggested(Reference Malavazos, Cereda and Morricone101, Reference Malavazos, Corsi and Ermetici102). Among the candidates secreted by the adipose tissue, the increase in IL-6, IL-8 and MCP-1 and the decrease in adiponectin are considered to be particularly important(Reference Malavazos, Cereda and Morricone101, Reference Malavazos, Corsi and Ermetici102). The studies of the pathophysiological links between adipokines and cardiovascular health can be illustrated by the analysis of the effects of adiponectin in rodents. Overexpression of adiponectin results in diminished size of the lesions observed following acute ischaemic myocardial infarction, increased angiogenic properties and reduced size of atheromatous plaques in the genetically predisposed apoE− / − mouse(Reference Ouchi, Kihara and Funahashi103).
Several inflammatory mediators produced by adipose tissue, such as CCL5, IL-1β and IL-8, as well as markers of oxidative stress, are increased in diabetic or glucose-intolerant patients, and the amelioration of hyperglycaemia by insulin therapy reduces circulating concentrations of these molecules(Reference Herder, Haastert and Muller-Scholze104, Reference Stentz, Umpierrez and Cuervo105). The increase in the concentrations of TNF-α, IL-6, IL-1β, IL-8, resistin and many other factors produced by macrophage activation could contribute to the deterioration of insulin sensitivity (Fig. 6)(Reference de Luca and Olefsky106). The precise relationship between the importance of macrophage and T-cell accumulation in adipose tissue depots, adipokine secretion and the modifications of insulin sensitivity needs to be further addressed in humans.

Fig. 6 Schematic representation of the role of adipose tissue inflammation in the initiation and maintenance of systemic insulin resistance. Reproduced with permission from de Luca & Olefsky(Reference de Luca and Olefsky106).
Macrophage accumulation is more abundant in the abdominal adipose tissue(Reference Cancello, Tordjman and Poitou65) than in the subcutaneous tissue, and this could explain some of the risks associated with the accumulation of intra-abdominal fat. For example, a relationship between the increase in macrophages in the abdominal adipose tissue and hepatic inflammation and fibrosis has been reported(Reference Cancello, Tordjman and Poitou65). In another study, the expression of the MCP-1 and colony-stimulating factor-1 genes and proteins was associated with macrophage accumulation in obese subjects(Reference Harman-Boehm, Bluher and Redel107). Since the abdominal adipose tissue is partly drained by the portal system, it cannot be excluded that some adipokines, together with high NEFA fluxes and hormones delivered by the adipose tissue, could contribute to the alteration of hepatic function observed in obese subjects, the mechanism of which needs to be better dissected.
Adipokines and weight loss
Even modest weight reduction improves the metabolic and cardiovascular risks associated with human obesity. Measures of endothelial activation also improve after weight reduction(Reference Berg and Scherer108–Reference Ziccardi, Nappo and Giugliano110). Many studies have shown that weight loss induced by a decrease in energy intake, and sometimes an increase in exercise, reduces systemic inflammation. A reduction in concentrations of numerous inflammatory molecules and endothelial risk factors, and an increase in adiponectin concentration have been observed during weight-loss programmes, and these are sometimes associated with improvement of insulin sensitivity(Reference Esposito, Pontillo and Di Palo111). Such changes have been described for CRP(Reference Heilbronn and Clifton112), IL-6(Reference Bastard, Jardel and Bruckert113), IL-18(Reference Esposito, Pontillo and Ciotola114), IL-1ra(Reference Meier, Bobbioni and Gabay26), PAI-1(Reference Bastard, Vidal and Jardel115), SAA(Reference Poitou, Viguerie and Cancello23, Reference Gomez-Ambrosi, Salvador and Rotellar116), cathepsin S(Reference Taleb, Cancello and Poitou117), matrix metalloproteinase-9(Reference Laimer, Kaser and Kranebitter118), soluble adhesion molecules (soluble intercellular adhesion molecule-1 (sICAM-1), soluble vascular cell adhesion molecule-1 (sVCAM-1))(Reference Ziccardi, Nappo and Giugliano110), tissue factor(Reference Kopp, Kopp and Steiner119), MIF(Reference Church, Willis and Priest120), MCP-1(Reference Christiansen, Richelsen and Bruun121), soluble receptors of TNF (sTNFR) and for eotaxin, an inflammatory factor implicated in asthma, another complication of obesity(Reference Vasudevan, Wu and Xydakis122). Weight loss induced by gastric bypass reduced the circulating concentrations of visfatin(Reference Krzyzanowska, Mittermayer and Krugluger123) and TNF-α(Reference Bastard, Hainque and Dusserre124, Reference Kopp, Kopp and Festa125). One study followed sixty obese patients during the course of weight loss induced by bariatric surgery and reported a reduction of 30 % of initial weight, a decrease in CRP, SAA, orosomucoid protein, IL-6, TNF-α and fibrinogen concentrations, and an increase in adiponectin concentration(Reference Poitou, Coussieu and Rouault126). After the surgery, the concentration of IL-6 dropped slowly while the concentrations of SAA and CRP dropped more quickly(Reference Poitou, Coussieu and Rouault126).
There was a significant modification in the expression of inflammatory genes in the subcutaneous adipose tissue of obese women following a hypoenergetic diet(Reference Clement, Viguerie and Poitou127). The expression of 100 genes linked to inflammatory processes was modified after 4 weeks (41 % increased and 59 % decreased). These genes belonged to at least twelve functional families including cytokines, the complement factor cascade, acute-phase proteins of inflammation, and molecules involved in cellular adhesion and in the remodelling of the extracellular matrix. The improvement of the inflammatory profile (at the level of gene expression) involved both the decreased expression of pro-inflammatory factors and the increased expression of anti-inflammatory factors such as IL-10 and IL-1ra. The modification of the inflammatory gene expression profile was very similar in subjects following bariatric surgery and was associated with a reduction of macrophage infiltration. In this study, the protein expression of IL-10 increased, suggesting a possible M1 to M2 (pro-inflammatory to anti-inflammatory) switch of macrophage phenotypes(Reference Cancello, Henegar and Viguerie31). Overall, the mitigation of the systemic inflammatory profile observed during weight loss is associated with modifications of adipose tissue inflammatory gene expression, and this may be linked with altered profiles of inflammatory protein secretion. The eventual consequence of this phenomenon on the local biology of the adipose tissue remains to be identified.
Chronic low-grade inflammation and insulin resistance
Experimental model systems in vitro
In obesity and the metabolic syndrome, key organs displaying increased insulin resistance are the liver, skeletal muscle, adipose tissue and the endothelium (Fig. 6). Experimental model systems in vitro have shown that hepatocytes as well as muscle cells, adipocytes and endothelial cells respond to exposure to the pro-inflammatory cytokines TNF-α, IL-6 and/or IL-1β with impaired insulin signalling(Reference de Luca and Olefsky128). Since these cytokines mediate their effects by binding to their cognate receptors, downstream components of the signalling cascade must interfere with insulin receptor function. Several such pathways have been identified (Fig. 7)(Reference de Luca and Olefsky128). Pro-inflammatory cytokine signalling usually activates the kinases IκB kinase β and C-jun N-terminal kinase 1, both of which in turn phosphorylate the Ser 307 residue of insulin receptor substrate (IRS)-1. This prevents the phosphorylation of tyrosine residues of IRS-1 by the insulin receptor and the downstream signalling cascade. TNF-α mediates insulin resistance also via the activation of p38 mitogen-activated protein kinase (MAPK) which interferes with the IRS–phosphatidylinositol 3-kinase–Akt pathway(Reference Li, Naples and Song129). Another mechanism involves cytokine-induced suppressor of cytokine signalling 1 and 3, which also prevent tyrosine phosphorylation of IRS proteins, by direct interference, or ubiquitylation and subsequent degradation(Reference Rui, Fan and Zhou130, Reference Ueki, Kondo and Kahn131). Induction of suppressor of cytokine signalling 3 by IL-6 occurs via the signal transducer and activator of transcription 3 (STAT3) mammalian target of rapamycin (mTOR) pathway(Reference Kim, Kim and Liu132).
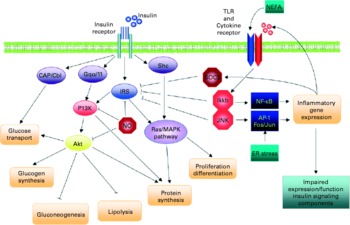
Fig. 7 Schematic representation of the direct interaction between inflammatory and insulin signalling pathways. The insulin signalling cascade branches into two main pathways. The PI3K-Akt pathway mediates insulin action on nutrient metabolism including glucose uptake. The Ras-mitogen-activated protein kinase (MAPK) pathway mediates the insulin's effect on gene expression, but also interacts with the PI3K-Akt pathway to control cell growth and differentiation. Activation of the insulin receptor leads to tyrosine phosphorylation of insulin receptor substrate (IRS)1, thereby initiating signal transduction. Stimulation of the NF-κB and activator protein-1 (AP-1) Fos/Jun inflammatory pathways results in the activation of serine kinases, Ikkβ and C-jun N-terminal kinase 1, which reduce the signalling ability of IRS1. Related negative regulators of IRS proteins include the suppressor of cytokine signalling proteins and NO, which are induced in inflammation, and promote IRS degradation. NO also reduces PI3K-Akt activity by nitrosylation of Akt. Reproduced with permission from de Luca & Olefsky(Reference de Luca and Olefsky128). TLR, Toll-like receptors.
In vivo models
The impairment of insulin signalling by TNF-α has also been observed in vivo after infusion of the cytokine into rodents(Reference Lang, Dobrescu and Bagby133, Reference Miles, Romeo and Higo134). A critical mediator downstream of TNF-α appears to be MIF, since mice with a disrupted MIF gene preserve normal insulin signalling(Reference Atsumi, Cho and Leng135). In this context, it is of interest that adipocytes are able to secrete MIF(Reference Skurk, Herder and Kraft33). The latter finding demonstrates that there are still substantial gaps in our understanding of the pro-inflammatory cytokine signalling cascade leading to insulin resistance. Most importantly, it remains unclear where other known regulators of insulin sensitivity fit into the chain of events. There is convincing evidence that reactive oxygen species (ROS) are critical to the effects of TNF-α on insulin signalling(Reference Houstis, Rosen and Lander136), and also that mitochondrial dysfunction is involved(Reference Nishikawa, Kukidome and Sonoda137). The impact of insulin on cellular metabolic activity, proliferation and differentiation can also be impaired by inflammatory mediators via an indirect pathway, i.e. by enhancing or suppressing the production of hormones that modulate cellular responses to insulin. These effects include the up- or down-regulation of the synthesis of resistin(Reference Li, Jiang and Xu138), leptin, adiponectin(Reference Arner139), lipocalin 2(Reference Yan, Barile and D'Agati140), osteopontin(Reference Nomiyama, Perez-Tilve and Ogawa141), and of insulin itself. When considering the subnanomolar systemic concentration of many pro-inflammatory mediators (see section ‘Obesity and low-grade inflammation’), it is possible that only a few of them contribute to the metabolic derangements seen in obesity and the metabolic syndrome.
A different situation emerges when paracrine effects of inflammatory mediators are considered. As described above, there is substantial local production of inflammatory mediators in organs affected by insulin resistance. Hepatocytes, adipocytes, muscle cells and the endothelium are sites of inflammatory mediator synthesis, but local activated macrophages appear to be the dominant site of synthesis and secretion, which leads to spillover into the general circulation(Reference Weisberg, McCann and Desai66, Reference Xu, Barnes and Yang67, Reference Clement and Langin142). Paracrine concentrations of inflammatory mediators are sufficient to induce insulin resistance(Reference de Luca and Olefsky128). Indeed, co-culture of adipocytes with macrophages caused impairment of insulin signalling(Reference Lumeng, Bodzin and Saltiel75). In addition to paracrine effects, it is conceivable that the functions of liver cells are affected by inflammatory mediators released from the abdominal adipose tissue because of their blood link.
Evidence supporting the link between inflammatory mediators and insulin resistance
In human subjects, the most direct approach to assess the contribution of low-grade inflammation to the development of insulin resistance and the metabolic syndrome is to analyse the consequences of anti-inflammatory pharmacotherapy. The longest experience is with the use of salicylates which are weak inhibitors of IκB kinase β and of serine phosphorylation of IRS proteins(Reference Kim, Kim and Fillmore143, Reference Yin, Yamamoto and Gaynor144). Early clinical trials with high doses of salicylates, notably aspirin, yielded both positive and negative effects on glycaemia and insulin resistance. Later studies have revealed that only very high doses are effective in improving glucose metabolism(Reference Shoelson, Lee and Yuan145). A randomised placebo-controlled pilot trial of salsalate treatment for 1 month in twenty non-diabetic obese individuals found decreased blood glucose and insulin responses to an oral glucose challenge consistent with improved insulin sensitivity(Reference Fleischman, Shoelson and Bernier146). A secondary analysis of a prospective multicentre observational study of 4905 adults with rheumatoid arthritis, of whom 1808 had taken hydroxychloroquine, indicated a reduced risk of diabetes in patients using this drug(Reference Wasko, Hubert and Lingala147). More specific anti-inflammatory intervention is possible through the use of recombinant proteins antagonising pro-inflammatory mediators. A first controlled double-blind trial was performed with daily injections of recombinant human IL-1ra for 13 weeks. This resulted in decreased glycated Hb levels and enhanced endogenous insulin production(Reference Larsen, Faulenbach and Vaag148). However, although there was a significant decrease in systemic CRP and IL-6 concentrations in response to anti-inflammatory treatment, there was no change in insulin resistance (homeostasis model assessment index and euglycaemic–hyperinsulinaemic clamp studies). It is difficult to judge the extent of inflammation persisting during therapy because absolute serum concentrations of CRP and IL-6 were not reported. There was no significant decrease in circulating TNF-α, MCP-1 or IL-8 concentrations, which indicates that there was no general down-regulation of pro-inflammatory cytokines. Another target-specific approach is the neutralisation of TNF-α by injections of recombinant antibodies or sTNFR. In animal models of insulin resistance, infusion of TNF-α antibodies has been reported to ameliorate insulin signalling(Reference Hotamisligil, Shargill and Spiegelman8, Reference Barbuio, Milanski and Bertolo149). In obese non-diabetic or diabetic individuals, several studies have observed improvement of insulin sensitivity after prolonged treatment with neutralising TNF-α antibodies(Reference Dominguez, Storgaard and Rask-Madsen150, Reference Tam, Tomlinson and Chu151), whereas other trials did not report such effects of TNF-α antibody injections, despite dampening of systemic inflammation(Reference Di Rocco, Manco and Rosa152). Possible explanations are that the recombinant antibodies do not reach sufficiently high concentrations in target tissues, or that TNF-α neutralisation is effective only in skeletal muscle tissue but not in adipose tissue as has been observed in rats(Reference Borst, Lee and Conover153). The overall conclusion is that results of studies of anti-inflammatory therapy generally support the concept of inflammatory mediators as contributors to the pathogenesis of insulin resistance, but have as yet not provided clear evidence of a critical pathogenic role of TNF-α or IL-1.
Postprandial inflammatory response
The foregoing discussion has dealt with chronic changes in concentrations of inflammatory mediators but a rise in inflammation also appears to take place acutely following meals. The postprandial inflammatory response lasts for only few (4–8) h but it recurs several times a day following eating. Although the postprandial inflammatory response has been known for several years(Reference Hansen, Sickelmann and Pietrowsky154), it is only recently that its probable importance in the generation of insulin resistance and atherosclerosis has been appreciated(Reference Plat, Jellema and Ramakers155–Reference O'Keefe, Gheewala and O'Keefe157). Several cells in the body associated with the innate immune system, including abdominal adipocytes and monocytes/macrophages, respond to acute postprandial elevation of several components of a meal by mounting a transient inflammatory response. The most efficient triggers of the postprandial inflammatory response appear to be TAG, SFA, oxysterols and glucose(Reference Napolitano and Bravo158–Reference Alipour, van Oostrom and Izraeljan161). The pathophysiological significance of a postprandial inflammatory response in causing insulin resistance, the metabolic syndrome and atherosclerosis is currently under investigation, and this response appears to play a much more crucial role than previously thought(Reference Burdge and Calder162).
Non-dietary factors influencing the magnitude of the postprandial inflammatory response
Body weight
Obesity is considered an important determinant of the magnitude of the postprandial inflammatory response(Reference Manning, Sutherland and McGrath163), perhaps being more important than any specific component of a meal inducing the response. The exaggerated postprandial inflammatory response of the obese is reversible upon reduction of body weight(Reference Plat, Jellema and Ramakers155, Reference Jellema, Plat and Mensink164).
Hyperglycaemia and type 2 diabetes
Patients with type 2 diabetes exhibit a higher postprandial inflammatory response than non-diabetics, irrespective of their body weight(Reference Esposito, Ciotola and Sasso165, Reference Nappo, Esposito and Cioffi166). The magnitude of the postprandial inflammatory response appears to correlate with the degree of insulin resistance(Reference Esposito, Ciotola and Sasso165).
Drugs
Certain medications including statins and angiotensin II receptor antagonists ameliorate the postprandial inflammatory response in obese patients(Reference Ceriello, Assaloni and Da167).
Pathophysiology of the postprandial inflammatory response
The daily influx of TAG, SFA, glucose and other food components initiates an acute innate immune (i.e. inflammatory) response that lasts for a few hours. Meals or food components may contain LPS which directly triggers systemic inflammation. Related to this effect, the absorptive process may allow translocation of LPS from gut bacteria into the systemic circulation(Reference Erridge, Attina and Spickett168). Meals may contain oxidised components which initiate oxidative stress and/or inflammatory responses upon absorption. Postprandial hyperglycaemia can suppress antioxidant capacity(Reference Ceriello, Bortolotti and Motz169) and thus its ability to curb an inflammatory reaction. Hyperglycaemia induces the production of free radicals which themselves initiate an inflammatory reaction. A six-transmembrane protein of prostate 2 (STAMP2) has been proposed as a major determinant of the postprandial inflammatory response(Reference Wellen, Fucho and Gregor170), acting to block activated inflammatory signalling pathways in adipocytes and possibly in macrophages. In vivo, feeding induces STAMP2 expression in visceral white adipose tissue(Reference Wellen, Fucho and Gregor170). Furthermore, the visceral tissue of STAMP gene knockout mice is resistant to insulin action(Reference Wellen, Fucho and Gregor170, Reference Abedini and Shoelson171).
Ageing and low-grade inflammation
Ageing is associated with complex changes in, and a dysregulation of, the immune system, including its inflammatory component. The ageing of the immune system, immunosenescence, has been suggested to be a consequence of continuous attrition caused by chronic antigenic overload(Reference Candore, Colonna-Romano and Balistreri172). Ageing is accompanied by a low-grade, chronic inflammatory state clearly shown by 2- to 4-fold increases in serum levels of several inflammatory mediators in older persons(Reference Krabbe, Pedersen and Bruunsgaard173). Studies have reported increased plasma/serum levels of the pro-inflammatory cytokine IL-6 in healthy subjects with advanced age (55–75 v. 26–54 years)(Reference Wei, Xu and Davies174), an increase of 0·016 pg/ml per year of life(Reference Hager, Machein and Krieger175) and a significant increase with overall age (from 20 to 102 years)(Reference Ferrucci, Corsi and Lauretani176), and in elderly diabetic subjects (65–80 years)(Reference Pedersen, Bruunsgaard and Weis177). Ageing is also associated with increased concentrations of TNF-α(Reference Paolisso, Rizzo and Mazziotti178, Reference Bruunsgaard, Andersen-Ranberg and Jeune179), CRP(Reference Ballou, Lozanski and Hodder180) and IL-1ra(Reference Ferrucci, Corsi and Lauretani176, Reference Roubenoff, Harris and Abad181). It is hypothesised that failure of anti-inflammatory mechanisms to neutralise inflammatory processes that are continuously triggered lifelong plays a role in chronic low-grade inflammation in the elderly(Reference Franceschi, Capri and Monti182). In line with this, it has recently been shown that ageing (two groups with a mean age of 77·9 and 102·5 years, respectively v. a group with a mean age of 43·5 years) is characterised by a profound reduction in anti-inflammatory lipoxin A4 levels(Reference Gangemi, Pescara and D'Urbano183).
The effect of ageing on the immune system, however, cannot be completely separated from the contribution of co-morbidity, medication use or malnutrition(Reference Ahluwalia184, Reference Lesourd185). Since several inflammatory markers act as disease markers, it is possible that some of the chronic low-grade inflammation patterns found in the elderly may be related to the presence of co-morbidities(Reference Ballou, Lozanski and Hodder180, Reference Jenny, Tracy and Ogg186). Interestingly, however, successful ageing (ageing without co-morbidities) has also been associated with chronic low-grade inflammation(Reference Krabbe, Pedersen and Bruunsgaard173). Other factors that may affect and modulate circulating levels of inflammatory mediators, including obesity, infections, physical activity, age-related decline in sex hormones and altered host–microbiota interaction at the gut level, may also be involved in the age-associated increase in low-grade inflammation(Reference Candore, Colonna-Romano and Balistreri172, Reference Bruunsgaard187–Reference Guigoz, Dore and Schiffrin189). Furthermore, high plasma levels of IL-6 (and TNF-α) in the elderly were associated with truncal fat mass(Reference Pedersen, Bruunsgaard and Weis177), suggesting that some of this effect might be mediated with age-associated increase in fat mass.
There is strong evidence that low-grade elevations of circulating inflammatory mediators are associated with the development of age-related conditions such as atherosclerosis, cognitive decline and frailty. This may in part reflect the inflammatory nature of these conditions which involve local or generalised inflammation (e.g. neuroinflammation in cognitive decline), with the increase in circulating concentrations of inflammatory mediators reflecting overspill from the inflammatory lesion(s). Additionally, the increased inflammatory burden could make a contribution to the ongoing pathology and to a worsening clinical situation. Increases in the levels of circulating TNF-α, IL-6, IL-2R and CRP are also strong predictors of all-cause mortality risk in longitudinal studies of several elderly cohorts(Reference Bruunsgaard, Ladelund and Pedersen190–Reference Volpato, Guralnik and Ferrucci193). However, whether increased inflammatory activity causes age-associated pathology or reflects the sum of ongoing pathological processes(Reference Krabbe, Pedersen and Bruunsgaard173, Reference Bruunsgaard, Pedersen and Pedersen194) remains uncertain. Survival analyses in studies from the USA and Europe with several populations (healthy, non-disabled, ≥ 65-year-old subjects(Reference Harris, Ferrucci and Tracy191), high-functioning subjects aged 70–79 years(Reference Reuben, Cheh and Harris192), disabled women aged ≥ 65 years(Reference Volpato, Guralnik and Ferrucci193) and relatively healthy 80-year-old people(Reference Bruunsgaard, Ladelund and Pedersen190)), however, show that effects of inflammatory mediators were independent of pre-existing morbidity and other traditional risk factors for death. This indicates that these inflammatory mediators influence pathological processes or act as very sensitive markers of subclinical disorders in the elderly(Reference Krabbe, Pedersen and Bruunsgaard173).
Exercise and low-grade inflammation
Influence of acute and regular physical activity and fitness on low-grade inflammation
The health benefits of a physically active lifestyle are well recognised. Physical inactivity and obesity are also increasingly recognised as modifiable behavioural risk factors for a wide range of chronic diseases, and in particular for CVD(Reference Pate, Pratt and Blair195). Physical fitness, physical exercise and physical activity are often used as interchangeable concepts, but it is important to point out the differences among these. Physical activity is any body movement that increases energy expenditure(Reference Caspersen, Powell and Christenson196). Self-reported data of physical activity are easy and feasible to ask in a questionnaire or interview in large populations but are a measurement subject to recall and reporting biases. Exercise is planned, structured and repetitive physical activity, while physical fitness is the capacity to perform physical activity, and makes reference to a full range of physiological and psychological qualities. To eliminate reporting bias that could be present in self-reported physical activity measurement, several studies have examined the relationship between cardiorespiratory fitness and inflammatory markers. Maximal oxygen consumption (VO2max) attained during a graded maximal exercise to voluntary exhaustion is considered as the single best indicator of cardiorespiratory fitness(Reference Shephard, Allen and Benade197). There are excellent reviews of the evidence addressing the influence of physical activity and fitness on low-grade inflammation from epidemiological studies as well as clinical trials on the general adult population(Reference Hamer198–Reference Panagiotakos, Kokkinos and Manios202), athletes(Reference Tomaszewski, Charchar and Przybycin203, Reference Northoff, Weinstock and Berg204), and in children and adolescents(Reference Wärnberg, Nova and Romeo205).
Acute v. regular exercise
IL-6 and other cytokines that are produced and released by skeletal muscles have been suggested to be involved in mediating the health-beneficial effects of exercise and to play important roles in the protection against diseases associated with low-grade inflammation. The following chain of events is based on observations by Pedersen and colleagues and has been excellently reviewed elsewhere(Reference Pedersen206–Reference Fischer, Berntsen and Perstrup208):
(1) Contracting skeletal muscle is a major source of circulating IL-6 in response to acute exercise. Plasma IL-6 increases in an exponential fashion with exercise and is related to exercise intensity, duration, the mass of muscle recruited and endurance capacity. During heavy exercise, such as a marathon, there is up to a 60-fold increase in plasma IL-6 concentration(Reference Ostrowski, Rohde and Zacho209), with the duration of the event explaining more than 50 % of the variation in concentration(Reference Fischer210). Interestingly, IL-6 shows a markedly lower response to acute exercise in trained subjects.
(2) Physiological concentrations of IL-6 stimulate the appearance in the circulation of the anti-inflammatory cytokines IL-1ra and IL-10 and inhibit the production of the pro-inflammatory cytokine TNF-α. The health benefits of long-term regular exercise are ascribed to the anti-inflammatory response elicited by an acute bout of exercise, which is partly mediated by muscle-derived IL-6.
(3) The anti-inflammatory effects of exercise may therefore offer protection against TNF-induced insulin resistance. Moreover, IL-6 stimulates lipolysis as well as fat oxidation. The increase in IL-6 at the end of exercise is responsible for the increased CRP levels during late recovery.
In response to regular physical activity, basal as well as post-exercise plasma concentrations of IL-6 will decrease by mechanisms that might include increased glycogen content, improved antioxidant capacity and improved insulin sensitivity. The lower concentrations of IL-6 in the circulation will subsequently result in lower CRP levels.
Few studies have prospectively examined the effect of exercise training on low-grade inflammatory status, and the data obtained from intervention studies are less consistent when compared with cross-sectional population studies. A lower number of subjects or a good physical condition in the start of some intervention studies may explain a part of this inconsistency. Nevertheless, two longitudinal studies in athletes show that regular training induces a reduction in CRP concentration(Reference Fallon, Fallon and Boston211, Reference Mattusch, Dufaux and Heine212). Conflicting findings exist in clinical trials that have involved exercise only. Several training interventions have not produced changes in basal IL-6 or CRP concentrations(Reference Hammett, Prapavessis and Baldi213–Reference Fischer, Plomgaard and Hansen218), while significant reductions in inflammatory markers have been observed following training without changes in BMI or body fat in elderly participants(Reference Kohut, McCann and Russell219, Reference Stewart, Flynn and Campbell220). The largest trial was performed in 652 sedentary healthy, young and middle-aged, white and black women and men in the HEalth, RIsk factors, exercise Training And Genetics (HERITAGE) Family Study(Reference Lakka, Lakka and Rankinen221). They were subjected to a 20-week standardised exercise training programme; there was no control group, and each subject served as its own control. A non-significant reduction in CRP concentration was consistent across all groups and varied between 1·2 and 2·2 mg/l. Considering that the over-time variation in CRP in healthy individuals with stable lifestyle is small(Reference Pearson, Mensah and Alexander222), the reduction, although not significant, could nevertheless reflect the true effect of exercise training. Further stratification according to basal CRP levels showed a reduction by about 1·3 mg/l in subjects with initial CRP levels above 3·0 mg/l.
Effects of exercise in elderly people
Elderly people have higher basal levels of inflammation independently of disease status, and a considerable number of studies have been carried out in this population to assess associations between physical activity and inflammatory markers(Reference Bruunsgaard, Ladelund and Pedersen190, Reference Elosua, Bartali and Ordovas223–Reference Jankord and Jemiolo230). Rather consistent inverse, BMI-independent, associations are found and the associations are suggested to be dose-dependent; the more physically active the person, the lower the inflammatory markers(Reference Fischer, Berntsen and Perstrup208, Reference Colbert, Visser and Simonsick224). Also subjects over 80 years of age show consistent inverse associations between inflammation and physical activity(Reference Bruunsgaard, Ladelund and Pedersen190). Functional fitness was inversely associated with IL-6 and IL-1ra concentrations (but not with CRP, TNF-α, IL-10 or IL-1β) in a prospective population-based study of 1020 participants aged 65 years and older(Reference Elosua, Bartali and Ordovas223, Reference Cesari, Penninx and Pahor231). Muscle strength was also evaluated in this study and low hand-grip strength was associated with high levels of CRP and IL-6(Reference Cesari, Penninx and Pahor231). Other studies have also shown a negative association of CRP, IL-6 and TNF-α with muscle strength(Reference Taaffe, Harris and Ferrucci228, Reference Visser, Pahor and Taaffe232).
Exercise intervention in elderly people or in patients with CVD shows consistent anti-inflammatory effects. After a 6-month individualised, supervised exercise programme for forty-three subjects at high risk of IHD, a 35 %, albeit non-significant, reduction in CRP concentration was observed. The subjects exercised for a mean of 2·5 (range 0·3–7·4) h/week(Reference Smith, Dykes and Douglas233). One reason for the lack of a significant effect despite the fairly large reduction in CRP concentration is the small size of the study. Another study reported a decrease in basal plasma IL-6 concentration after aerobic training in patients with coronary artery disease(Reference Goldhammer, Tanchilevitch and Maor234). A randomised trial of thirty-nine patients with intermittent claudication demonstrated that both serum CRP and SAA concentrations were significantly reduced after 3 and 6 months of supervised exercise compared with controls(Reference Tisi, Hulse and Chulakadabba235). In a relatively large intervention study of exercise training on cardiac rehabilitation patients, the median CRP concentration was reduced by 41 %, while CRP concentrations did not change in subjects who did not exercise(Reference Milani, Lavie and Mehra236). Again, the exercise training seemed to be more effective in those with the highest initial CRP concentrations, independently of changes in body weight or percentage of body fat, indicating that baseline levels of low-grade inflammation may be an important factor.
Studies in patients with diabetes(Reference McGavock, Mandic and Vonder Muhll237) or the metabolic syndrome(Reference Aronson, Sella and Sheikh-Ahmad238, Reference Aronson, Sheikh-Ahmad and Avizohar239) have consistently demonstrated inverse associations between fitness and inflammation, independently of fatness. In one study, the independent associations of fitness were in fact more prominent among metabolic syndrome patients compared with healthy participants(Reference Aronson, Sella and Sheikh-Ahmad238, Reference Aronson, Sheikh-Ahmad and Avizohar239).
Effects of exercise in middle-aged and younger adults and in children
Several studies of large population cohorts, such as the British Regional Heart Study(Reference Wannamethee, Lowe and Whincup225), the Greek ATTICA study(Reference Panagiotakos, Pitsavos and Chrysohoou240), the Third National Health and Nutrition Examination Survey (NHANES)(Reference Abramson and Vaccarino241, Reference King, Carek and Mainous242), the Men's Health Professionals’ Follow-Up Study(Reference Pischon, Hankinson and Hotamisligil243), the Nurses’ Health Study(Reference Pischon, Hankinson and Hotamisligil243) and the Women's Health Study(Reference Mora, Lee and Buring244), provide evidence for an inverse, independent dose–response relationship between plasma CRP concentration and level of physical activity in both men and women, but the consistency is less than in elderly subjects, or in disease states. In contrast, the associations found between self-reported physical activity and TNFR1, TNFR2, IL-6 and CRP concentrations in a study including healthy men from the Men's Health Professionals’ Follow-Up Study and healthy women from the Nurses’ Health Study(Reference Pischon, Hankinson and Hotamisligil243) were no longer significant when adjusting for BMI and leptin. Thus, the effect of physical activity on circulating markers of low-grade inflammation appears to be mediated by weight loss. In another study in healthy men and women, BMI, but not previous year or current physical activity, predicted CRP concentration(Reference Rawson, Freedson and Osganian245). Similarly, a cross-sectional study(Reference Verdaet, Dendale and De246) in men found no relationship between leisure-time physical activity and CRP, fibrinogen and SAA concentrations, after correction for BMI and current smoking status. CRP levels in 2120 Finns were associated with obesity indices and physical activity among both sexes(Reference Raitakari, Mansikkaniemi and Marniemi247); in multivariate analyses, the determinants of CRP concentration included obesity and smoking in men, and obesity, oral contraceptive use and physical activity in women. The study showed that about one in three of healthy women who used oral contraceptives had a CRP concentration exceeding 3 mg/l, which should be taken into account when studying younger females. Cross-sectional studies in men from the Aerobics Center Longitudinal Study have demonstrated that cardiorespiratory fitness levels are inversely associated with CRP concentration and also the prevalence of elevated CRP concentrations(Reference Church, Barlow and Earnest248). Analyses with fibrinogen and white blood cell count showed similar results(Reference Church, Finley and Earnest249). The competing effect of weight and fitness (assessed by submaximal graded exercise treadmill testing) on cardiorespiratory fitness levels was studied in the NHANES, which included 2112 US adults without previously diagnosed CVD(Reference Diaz, Player and Mainous250). Both fitness and BMI were independently associated with increased fasting insulin and CRP concentrations. However, when patients with low, moderate and high fitness were further stratified as normal, overweight or obese, weight remained significantly associated with CRP, but fitness did not. This study concludes that ‘fat but fit’ subjects require weight-loss interventions to improve their CRP levels. Future interventions should emphasise weight control, even for those with high cardiorespiratory fitness.
In disease-free young populations, studies have assessed the interaction between inflammatory markers (CRP, IL-6 and TNF-α), physical activity or cardiorespiratory fitness and fatness(Reference Cook, Mendall and Whincup47, Reference Isasi, Deckelbaum and Tracy251–Reference Wärnberg258). Organised leisure-time exercise (assessed by a questionnaire) in children has shown negative correlations with serum IL-6 concentrations, independently of adiposity and fat localisation(Reference Platat, Wagner and Klumpp256), and in 10-year-old children, a borderline significant negative association was observed between CRP and self-reported physical activity, independently of ponderal index(Reference Cook, Mendall and Whincup47). US children and young adults (aged 6–24 years) from the Columbia University BioMarkers Study showed an inverse correlation between cardiovascular fitness and CRP concentration but only in boys, which remained after adjustment of confounders including BMI(Reference Isasi, Deckelbaum and Tracy251). Only one study has used accelerometry (an objective measure of total physical activity compared with leisure-time physical activity or exercise) instead of questionnaires as well as cardiovascular fitness(Reference Ruiz, Ortega and Warnberg257). In this study of 9-year-old Swedish children, total physical activity was unrelated to CRP, fibrinogen, C3 or C4 concentrations, but exercise was. Nevertheless, once body fat was entered into the regression models, no associations with cardiovascular fitness or physical activity and the inflammatory markers measured were observed(Reference Ruiz, Ortega and Warnberg257). Similarly, no associations were found between cardiorespiratory fitness or self-reported physical activity and CRP concentration in 12-year-old healthy Welsh children(Reference Thomas, Baker and Graham259).
CRP, C3 and ceruloplasmin (but not C4) concentrations were negatively associated with muscle strength after controlling for sex, age, pubertal status, weight, height, socio-economic status and cardiorespiratory fitness, but did not remain when adjusting for body fat. Nevertheless, when stratifying according to overweight status, CRP (but not C3, C4 or ceruloplasmin) concentration was associated with muscle strength in overweight, but not in normal-weight, adolescents after controlling for potential confounders, including body fat and fat-free mass(Reference Ruiz, Ortega and Warnberg260).
Conclusions for effects of physical activity and fitness on low-grade inflammation
Most research on this topic hypothesised that the association between fitness and inflammatory factors is independent of fatness. Given that physical activity and obesity are often inversely related, it is not clear as to whether the anti-inflammatory health benefits of a physically active lifestyle are due to exercise per se or result from favourable changes in body composition. Related anti-inflammatory effects could be mediated by increased insulin sensitivity and/or improved concentrations of HDL-cholesterol, ROS or endothelial function, which all demonstrate anti-inflammatory actions(Reference Dandona, Aljada and Chaudhuri261), and are related to both body fat and exercise. A systematic review addressed whether fitness or fatness has the greatest impact on inflammatory factors(Reference Hamer198). The review concluded that both fitness and fatness are associated with systemic inflammatory status, although the relative contributions of both may be dependent on age, disease status and sex. These determinants do most probably involve a strong background of low-grade inflammatory status, which consistently is shown to determine any possible inverse association. Although increasing physical activity may be an effective therapy for weight loss and may also emerge as a promising treatment for reducing overall inflammation, the magnitude of the effect to produce clinically meaningful results in the general population requires further research(Reference Nicklas, You and Pahor200). Nevertheless, exercise is uniquely positioned to reduce inflammation, and even small non-significant reductions in CRP levels may contribute to clinical benefits by reducing cardiovascular and metabolic risk(Reference Pearson, Mensah and Alexander222).
A consideration of different approaches to identify relationships between diet and its components and markers of chronic low-grade inflammation
The following sections review the effects of dietary factors, including dietary patterns, whole foods, individual nutrients and other bioactive components on the markers of low-grade inflammation described above. Due to the physiological complexities detailed above, assessment of effects requires careful attention to treatment interventions and study designs, in the context of the endpoints described. Here, important aspects of study design are briefly mentioned.
Epidemiological studies
Epidemiological studies, where available, are discussed for each of the dietary factors. As mentioned above, body-weight changes and exercise may have profound effects on biomarkers of low-grade inflammation; therefore, adjustments for BMI and activity level are critical. An accurate assessment of the analysed dietary parameter is also necessary. Although difficult to rectify, it is important to consider cultural differences in food consumption habits, for example differences in coffee preparation procedure and brew strength between the USA and Europe. Attention should also be paid to the assessment of dietary patterns. For example, the Mediterranean diet has been variously defined and scored as inclusive or exclusive of fish, poultry, dairy, eggs, moderate alcohol consumption and ratio of monounsaturated:saturated fat. However, it is not clear whether these differences in scoring would result in alternative conclusions being drawn, and as such, this body of literature is best viewed in totality.
Intervention studies
Intervention studies, where available, are also discussed for each of the dietary factors. The studies presented here typically fall into three design categories: (1) chronic dietary interventions in individuals with some degree of existing low-grade inflammation, based on changes in markers measured in fasting blood, sometimes in the context of weight loss; (2) acute dietary interventions in which the acute effect of a putative anti-inflammatory dietary treatment is assessed against some background level of low-grade inflammation; (3) challenge studies in which inflammation is induced by either a dietary or exercise challenge, in the presence or absence of a putative anti-inflammatory dietary treatment. As with epidemiological studies, body weight and activity levels require careful control and monitoring because of their possible impact on low-grade inflammation. Chronic interventions are most commonly either parallel-arm designs or cross-over designs, and may be most relevant since they directly evaluate the effect of the dietary component on low-grade inflammation. However, acute intervention and postprandial challenge studies may also provide valuable insight, and postprandial inflammation has recently been hypothesised to play an aetiological role in the progression of CVD. Additionally, between-subject variations in biomarkers of the inflammatory response can be controlled for statistically in cross-over designs. In contrast to epidemiological studies, which often have a large sample size and may evaluate outcomes over a long duration, intervention studies are most often relatively small and of short duration (hours for acute studies; weeks to months for chronic studies). Small studies can limit power to identify significant effects. Another difficulty with intervention studies can be compliance among subjects; although an ideal study design would include actions to ensure compliance and would monitor this, such approaches are not always considered. Lack of compliance may limit the effectiveness of an intervention.
Dietary patterns and low-grade inflammation
For the purposes of this article, low-grade inflammation was defined as elevated circulating concentrations of pro-inflammatory cytokines, acute-phase proteins and adhesion molecules, and low circulating concentrations of adiponectin.
Eating patterns
Studies on diet and disease have traditionally examined the associations of individual nutrients, foods or food groups with risk factors and health outcomes. However, this approach has certain limitations: many nutrients are highly correlated, and have synergistic or interactive effects; examination of nutrients or foods singly may not provide enough statistical power due to the small effect size; and the possibility of finding significant associations by chance alone, due to multiple testing, is large. In response to the challenges of the traditional approach to understanding diet–disease relationships, more recently, nutrition epidemiologists have studied dietary or eating patterns that examine combinations of foods and nutrients, in relation to health and disease(Reference Moeller, Reedy and Millen262, Reference Newby, Muller and Hallfrisch263). Dietary pattern research is generally based on two kinds of methods: a priori using diet scores; or a posteriori using data-driven techniques such as factor analysis and cluster analysis(Reference Michels and Schulze264–Reference Hu266).
Hypoenergetic diet
One diet-dependent but apparently quite non-specific way of decreasing low-grade inflammation is energy restriction(Reference Holloszy and Fontana267). Weight loss is accompanied by decreased concentrations of circulating mediators of inflammation, such as CRP, TNF-α, IL-6 and sICAM-1(Reference Plat, Jellema and Ramakers155, Reference Kolb and Mandrup-Poulsen268–Reference Madsen, Rissanen and Bruun270), although it is difficult to dissect whether this effect is due to the weight loss per se or to the nature of the diet used to induce weight loss. On the other hand, it seems likely that reduced secretion of pro-inflammatory mediators from adipocytes or activated macrophages of adipose tissue contributes to the effect of weight loss(Reference Cancello, Henegar and Viguerie31, Reference Curat, Wegner and Sengenes36, Reference Cancello, Tordjman and Poitou65, Reference Xu, Barnes and Yang67, Reference Clement and Langin142). However, energy restriction itself may also play an anti-inflammatory role, with key mediators of the effect being proteins of the sirtuin and Forkhead box, sub-group O (FoxO) families which are induced/activated during states of limited energy supply. The sirtuins are NAD+-dependent deacetylases of substrates ranging from histones to transcriptional regulators. As a consequence, metabolic efficiency is improved, cell defences against stress are strengthened and inflammatory activities are dampened, notably by decreasing the activation of NF-κB(Reference Guarente and Picard271–Reference Salminen, Ojala and Huuskonen274). FoxO proteins are transcription factors which regulate the expression of genes involved in energy homeostasis, cell survival and inflammatory responses including NF-κB(Reference Salminen, Ojala and Huuskonen274–Reference Nakae, Oki and Cao278). Early studies have shown that reduced energy intake is paramount compared with the nature of low-energy food, i.e. decreased concentrations of inflammatory markers are observed with a low-energy, fat-rich as well as with a low-energy, carbohydrate-rich diet(Reference Sharman and Volek279). However, it is conceivable that some dietary components are better regulators of sirtuin or FoxO activity than others. Resveratrol, present in red wine, was found to directly or indirectly interact with sirtuins and promote their deacetylase activity. This led to increased lifespan in model organisms and in animal models(Reference Baur, Pearson and Price280, Reference Baur and Sinclair281). It is likely that dietary components will be identified that mimic or counteract the anti-inflammatory effects of energy restriction.
Mediterranean diet
The term Mediterranean diet refers to a traditional dietary pattern characteristic of many parts of Greece, Southern Italy, Southern Spain and elsewhere in the Mediterranean region. The traditional Mediterranean diet is rich in fruit, vegetables, whole-grains, legumes (beans), nuts, fish and low-fat dairy products, with moderate consumption of wine, and whose principal source of fat is olive oil(Reference Dai, Miller and Bremner282–Reference Trichopoulou, Costacou and Bamia286). Most studies have assessed the adherence to the Mediterranean diet by assigning a score in relation to the consumption of these foods. Others(Reference Esposito, Marfella and Ciotola284, Reference Fung, McCullough and Newby285) have used modified versions of this score by not considering certain food groups, i.e. dairy products.
Observational studies have examined the association of the Mediterranean diet with inflammatory markers in healthy persons(Reference Dai, Miller and Bremner282, Reference Chrysohoou, Panagiotakos and Pitsavos283, Reference Fung, McCullough and Newby285), and they generally report inverse correlations. In a recent study(Reference Dai, Miller and Bremner282) investigating psychological, behavioural and biological risk factors for subclinical CVD in 345 middle-aged male twins, an inverse association between adherence to the Mediterranean diet and inflammation, as measured by plasma IL-6 concentration, was noted. This association was independent of several known cardiovascular risk factors, and persisted when twins within pairs were compared, suggesting that the results were not confounded by shared environmental and genetic factors. Although a marginal relationship between CRP concentration and the Mediterranean diet was observed, this association was no longer significant when other cardiovascular risk factors were considered in the models. It is likely that IL-6 is a more sensitive marker of chronic low-grade inflammation and that CRP reflects a more downstream effect associated with IL-6. In a subsample of the Nurses’ Health Study(Reference Fung, McCullough and Newby285), a Mediterranean diet index score was inversely associated with markers of inflammation (circulating IL-6 and CRP) as well as markers of endothelial dysfunction (the adhesion molecules sICAM-1, sVCAM-1 and soluble E-selectin (sE-selectin)); these associations persisted upon adjustment for traditional CVD risk factors. Similar findings were reported in the ATTICA study, involving 1514 men and 1528 women; specifically, subjects with greater adherence to the Mediterranean diet (those in the highest tertile) had 17 % lower IL-6 and 20 % lower CRP concentrations, compared with those in the lowest tertile in analyses that adjusted for other cardiovascular risk factors(Reference Chrysohoou, Panagiotakos and Pitsavos283). Although a marginal association was noted between TNF-α and the Mediterranean diet, it was not significant in adjusted models(Reference Chrysohoou, Panagiotakos and Pitsavos283). In another observational study with obese subjects (625 men and 712 women with abdominal adiposity), those with high CRP levels (>3 mg/l) were less likely to adopt the Mediterranean diet(Reference Pitsavos, Panagiotakos and Tzima287). The authors reported that adoption of the Mediterranean diet in conjunction with moderate physical activity was associated with a reduced likelihood of having high CRP levels by 72 %, highlighting the potential importance of the Mediterranean diet in diminishing inflammation. In subjects at high risk for CVD, those with diabetes or with multiple CHD risk factors, however, no association between the Mediterranean diet and CRP or adhesion molecule (sICAM-1 and sVCAM-1) concentrations was seen(Reference Salas-Salvado, Garcia-Arellano and Estruch288). However, a significant relationship between higher consumption of some typical Mediterranean diet components (cereals, fruit, nuts and virgin olive oil) and circulating inflammatory markers (IL-6) and markers of endothelial function was noted in these at-risk subjects.
Few intervention studies have been conducted to examine the effect of consuming the Mediterranean diet on markers of low-grade inflammation. In two cross-over studies involving short-term interventions (1–3 months) with healthy subjects, differential effects of the Mediterranean diet on markers of inflammation (and endothelial dysfunction) were observed. In the study by Ambring et al. (Reference Ambring, Johansson and Axelsen289), healthy subjects received their typical Swedish diet for about 4 weeks, and a Mediterranean-inspired diet for about 4 weeks in a randomised cross-over design, with 4 weeks of washout in-between. A marked reduction in the number of leucocytes, including monocytes, neutrophils and lymphocytes, and in the number of platelets after 4 weeks of the Mediterranean diet was noted, suggesting a lower inflammatory activity than during the Swedish diet period. On the other hand, IL-6 and CRP concentrations did not change with the Mediterranean diet; the authors speculated that the study may not have been powered to detect those effects. In another study(Reference Bellido, Lopez-Miranda and Perez-Martinez290), twenty healthy young males were provided three dietary interventions, each lasting 4 weeks. First, all subjects consumed a diet high in saturated fat, next they were randomly assigned to either of the two intervention diets: a Mediterranean-style diet (MUFA-enriched diet, 22 % energy from MUFA) or a low-fat, high-carbohydrate diet ( < 30 % energy from fat and 55 % from carbohydrates). LDL was isolated and oxidised. Oxidised LDL from subjects on either the Mediterranean diet or the high-carbohydrate diet decreased TNF-α-induced VCAM-1 and E-selectin expression in human umbilical endothelial cells in vitro. A consistent decline in inflammatory markers has been observed in intervention studies involving obese subjects or those with the metabolic syndrome. In a randomised controlled study with 120 pre-menopausal obese women, the effects of a multidisciplinary approach (aiming at 10 % weight reduction with a combination of a low-energy, Mediterranean-style diet and increased physical activity) were evaluated compared with a control group (given general information about healthy choices and exercise)(Reference Esposito, Pontillo and Di Palo111). The intervention group received regular sessions (18 over a 2-year period) with a nutritionist to ensure compliance. Significant reduction in several markers of inflammation (CRP, IL-6 and IL-18) and an increase in adiponectin concentration were noted in the Mediterranean diet group compared with the control group. In a randomised controlled trial lasting 2 years, 180 subjects with the metabolic syndrome were assigned either to a Mediterranean-style diet (instructions were provided on increasing daily consumption of whole grains, fruit, vegetables, nuts and olive oil) or to a control group (prudent diet, with same macronutrient composition as the Mediterranean diet)(Reference Esposito, Marfella and Ciotola284). After 2 years, body weight decreased more in the intervention group and inflammatory markers (CRP, IL-6, IL-7 and IL-18) decreased and endothelial function improved, compared with the control group. Interestingly, even after controlling for weight loss, the inflammatory markers declined more in subjects following the Mediterranean-style diet. In other related studies involving individuals with the metabolic syndrome, a consistent reduction in CRP concentration in the intervention group receiving the Mediterranean diet has been shown(Reference Esposito, Ciotola and Giugliano291, Reference Esposito, Ciotola and Giugliano292). In the Prevención con Dieta Mediterránea study, 772 asymptomatic subjects at high cardiovascular risk (diabetes or more than three CHD risk factors) were randomly assigned to a low-fat diet or one of two Mediterranean diets(Reference Estruch, Martinez-Gonzalez and Corella293). Those allocated to the Mediterranean diets received nutritional education and either free virgin olive oil, or free nuts for 3 months. Both Mediterranean diets were beneficial in terms of significant reductions in serum IL-6, sICAM-1 and sVCAM-1 concentrations, while CRP concentration was reduced only in the Mediterranean diet supplemented with olive oil. Taken together, the results from these often large intervention studies strongly suggest that Mediterranean diets can lead to reductions in chronic low-grade inflammation and improvement in endothelial function, thereby offering cardioprotective effects.
Vegetarian diets
Observational studies have compared vegetarian with non-vegetarian diets in healthy subjects in relation to inflammatory markers. Dietary patterns consistent with vegetarianism were associated with lower concentrations of markers of chronic low-grade inflammation and endothelial function when compared with non-vegetarian diets(Reference Krajcovicova-Kudlackova and Blazicek294–Reference Purschwitz, Rassoul and Reuter296). In one study comparing a group of thirty Taoist adults who had been vegetarian for 5–55 years (average 22 years) with a group of thirty age- and sex-matched non-Taoist adults consuming a non-vegetarian diet, lower CRP concentrations were noted in the former group(Reference Szeto, Kwok and Benzie295). Specifically, plasma CRP concentration averaged 0·77 and 1·30 mg/l in vegetarians and non-vegetarians, respectively. Another study with a similar design comparing vegetarians with non-vegetarians also found that the average CRP concentration was significantly lower in the vegetarian group (0·72 v. 1·62 mg/l)(Reference Krajcovicova-Kudlackova and Blazicek294). Similarly, lower levels of the adhesion molecules sICAM-1 and sE-selectin have been reported in those following a vegetarian v. a non-vegetarian diet(Reference Purschwitz, Rassoul and Reuter296). Thus, these cross-sectional studies suggest that a vegetarian-style diet can lead to lower chronic low-grade inflammation than an omnivorous diet. However, it is important to recognise that vegetarians may differ from non-vegetarians in aspects of lifestyle other than diet such as physical activity, smoking behaviour and socio-economic class. Studies considering vegetables and fruits are described in section ‘Vegetables and fruits’.
Eating patterns
The healthy eating index (HEI) was developed by the US Department of Agriculture, based on the Dietary Guidelines for Americans and the Food Guide Pyramid(Reference Kennedy, Ohls and Carlon297). It has ten subcomponents: grains, fruits, vegetables, dairy, meats, fats, saturated fat, cholesterol, Na and dietary variety. Individuals are assigned scores for each of the ten components (from 0 to 10) based on their typical intakes; the maximum value for the HEI being 100. Recently, the HEI was revised as the Alternate HEI (AHEI) to focus on healthier items in the Food Guide Pyramid food groups(Reference McCullough, Feskanich and Stampfer298) such as protein source, ratio of polyunsaturated:saturated fats and cereal fibre, as well as cis- v. trans-fat. Additionally, moderate alcohol consumption and long-term multivitamin use are also considered in the AHEI. The Diet Quality Index (DQI) is a composite score of an overall healthy diet that reflects an individual's adherence to the eight diet and health recommendations of the National Academy of Science. The revised DQI (DQI-R) is based on similar guidelines but also includes Fe and Ca intakes(Reference Haines, Siega-Riz and Popkin299).
Using the data from NHANES III on a representative sample of the US population, Ford et al. (Reference Ford, Mokdad and Liu300) found a negative correlation between the HEI and CRP concentration after adjustment for several CVD risk factors including BMI and waist:hip ratio; when stratified by sex this finding was significant in women only. The authors noted that this association was primarily driven by grain consumption. The authors speculated that because HEI score was determined based on dietary intake data collected by a 24 h recall, the possibility of misclassification of individuals with respect to HEI status could have attenuated the associations. In a study by Fung et al. (Reference Fung, McCullough and Newby285), the association of several diet-quality scores, such as the HEI, AHEI, DQI-R and an alternate Mediterranean diet index with various markers of inflammation, was evaluated in a subsample of the Nurses’ Health Study (n 690 healthy women). The key findings were that the AHEI and alternate Mediterranean diet index scores were negatively associated with CRP, IL-6, sE-selectin and sICAM-1 concentrations, and that these associations persisted upon adjustment for potential confounding variables including BMI. In contrast, the HEI and DQI-R did not correlate with any of the inflammatory markers when potential confounders were taken into consideration in the regression models(Reference Fung, McCullough and Newby285). A more recent study evaluated the impact of the AHEI among 1922 women from the Nurses’ Health Study (62 % of whom were overweight) who had no history of diabetes or CVD on plasma inflammatory marker concentrations(Reference Fargnoli, Fung and Olenczuk301). After adjustment for age and energy intake, women with the highest adherence to the AHEI had 24 % higher median total adiponectin and 32 % higher median high-molecular-weight adiponectin concentrations, as well as 16 % lower resistin, 41 % lower CRP and 19 % lower sE-selectin concentrations than did women with the lowest adherence to the AHEI. These associations remained significant after adjustment for potential confounders. Inverse associations between the AHEI and CRP, sTNFR2, IL-6, sICAM-1 and sVCAM-1 concentrations were evident, but they did not remain significant after adjustment for BMI. In a small cross-sectional study involving 114 ‘apparently healthy’ overweight or obese postmenopausal women, Boynton et al. (Reference Boynton, Neuhouser and Wener302) found little evidence of an association between dietary quality, as measured by the HEI and DQI, and markers of inflammation (CRP and SAA). Marginal associations were noted between the DQI and these inflammatory markers. These associations were, however, attenuated and no longer significant, after adjusting for adiposity (percentage of body fat or BMI), suggesting that the decrease in CRP or SAA concentration seen with a higher-quality diet is most probably mediated by obesity. The authors speculated that consuming a healthier diet may lead to decreased adiposity, which in turn could result in less low-grade inflammation.
Several studies have examined the relationship between consuming a ‘prudent diet’ and markers of low-grade inflammation. In one study, in 732 healthy women from the Nurses’ Health Study aged 43–69 years, a prudent pattern was characterised by higher intakes of fruit, vegetables, legumes, fish, poultry and whole grains, and a Western pattern was characterised by higher intakes of red and processed meats, sweets, desserts, French fries and refined grains(Reference Lopez-Garcia, Schulze and Fung303). The prudent pattern was inversely associated with plasma concentrations of CRP and sE-selectin after adjustment for age, BMI, physical activity, smoking status and alcohol consumption. The Western pattern showed a positive relationship with CRP, IL-6, sE-selectin, sICAM-1 and sVCAM-1 after adjustment for all confounders except BMI; with further adjustment for BMI, the coefficients remained significant for CRP, sE-selectin, sICAM-1 and sVCAM-1.Using data from the Nurses’ Health Study, Schulze et al. (Reference Schulze, Hoffmann and Manson304) identified a dietary pattern that was significantly associated with increased concentrations of CRP, IL-6, sTNFR2, sE-selectin, sICAM-1 and sVCAM-1. This pattern, which was high in sugar-sweetened soft drinks, refined grains, diet soft drinks and processed meat but low in wine, coffee, cruciferous vegetables and yellow vegetables, was also associated with an increased risk of diabetes. Most recently, Hoebeeck et al. (Reference Hoebeeck, Rietzschel and Langlois305) evaluated the relationship between adherence to food-based dietary guidelines and inflammatory markers among 2524 healthy Belgian men and women aged 35–55 years. The dietary index consisted of three subscores (dietary quality, diversity and equilibrium) according to adherence to the Flemish food-based dietary guidelines, using data from a semi-quantitative FFQ. Higher dietary scores were inversely associated with IL-6 concentration and leucocyte numbers in the bloodstream. Nettleton et al. (Reference Nettleton, Steffen and Mayer-Davis306) examined relationships between dietary patterns and markers of inflammation and endothelial activation among 5089 non-diabetic participants in the Multi-Ethnic Study of Atherosclerosis. In this study, four dietary patterns were derived by using factor analysis. The fats and processed meats pattern (fats, oils, processed meats, fried potatoes, salty snacks and desserts) was positively associated with CRP and IL-6 concentrations. The beans, tomatoes and refined grains pattern (beans, tomatoes, refined grains and high-fat dairy products) was positively related to sICAM-1 concentration. In contrast, the whole grains and fruit pattern (whole grains, fruit, nuts and green leafy vegetables) was inversely associated with CRP, IL-6 and sICAM-1 concentrations, while the vegetables and fish pattern (fish and dark-yellow, cruciferous and other vegetables) was inversely related to IL-6 concentration. There are few such studies using data from outside of the USA or Europe. A cross-sectional study of 486 healthy Iranian women aged 40–60 years identified dietary patterns by factor analysis and related these to circulating markers of inflammation(Reference Esmaillzadeh, Kimiagar and Mehrabi307). The healthy dietary pattern (high in fruits, vegetables, tomatoes, poultry, legumes, tea, fruit juices and whole grains) was inversely related to plasma CRP, sE-selectin and sVCAM-1 concentrations after controlling for potential confounders; with further adjustment for BMI and waist circumference, the associations remained significant for CRP and sVCAM-1. In contrast, the Western dietary pattern score (high in refined grains, red meat, butter, processed meat, high-fat dairy, sweets and desserts, pizza, potatoes, eggs, hydrogenated fats and soft drinks) was positively related to CRP, SAA, IL-6, sICAM-1 and sVCAM-1 concentrations; however, after additional control for BMI and waist circumference, the associations remained significant only for SAA and IL-6.The traditional dietary pattern (high in refined grains, potatoes, tea, whole grains, hydrogenated fats, legumes and casserole) was positively associated with the plasma IL-6 concentration after controlling for BMI and waist circumference. Nanri et al. (Reference Nanri, Yoshida and Yamaji308) investigated the relationship between dietary patterns and circulating CRP concentration in 7802 Japanese men and women with CRP <3 mg/l. The dietary patterns were derived from principal component analysis of the frequency of consumption of forty-nine food items ascertained by the FFQ. The following four dietary patterns were identified: healthy, high-fat, seafood and Westernised breakfast patterns. The healthy dietary pattern, characterised by high intakes of vegetables, fruits, soya products and fish, was inversely related to CRP concentrations, even after adjustment for age, BMI, smoking, alcohol consumption and physical activity in both men and women. Neither the high-fat dietary pattern nor the Westernised breakfast pattern was related to CRP concentrations. Most recently, the relationship between two dietary patterns (Healthy and Western), which were derived by principal component analysis using data collected by a FFQ from subjects in the Atherosclerosis Risk in Communities Study, and markers of inflammation has been described(Reference Nettleton, Matijevic and Follis309). The measures of inflammation were quantified by flow cytometry in fresh whole blood from 1101 white adults. After multivariable adjustment, monocyte LPS receptor (CD14), monocyte TLR-2 and platelet glycoprotein IIb (CD41) showed inverse associations with the healthy dietary pattern. In contrast, the Western dietary pattern was positively associated with CD41 and platelet–granulocyte aggregates. Taken together, these studies suggest that healthy eating patterns are associated with lower concentrations of markers of chronic low-grade inflammation.
Whole foods
Whole grains/refined grains
Published studies have so far investigated a narrow and low range of whole grain intakes, which limits the interpretation of associations between whole grain intake and markers of low-grade chronic inflammation. Observational studies (Table 3)(Reference Ford, Mokdad and Liu300, Reference Jensen, Koh-Banerjee and Franz310–Reference Qi, van Dam and Liu314) including data from NHANES III have suggested that a high intake of whole grain is inversely associated with plasma CRP concentration (quintile (Q) 1 < 3·5 servings/d, Q5>9·7 servings/d)(Reference Ford, Mokdad and Liu300). In contrast, Jensen et al. (Reference Jensen, Koh-Banerjee and Franz310) reported no association between a moderate whole grain intake (Q1 8·2 g/d, Q5 43·8 g/d) and markers of inflammation (CRP, IL-6 and fibrinogen) in the Health Professionals’ Follow-Up Study. However, bran intake correlated inversely with CRP concentration and germ intake with IL-6 concentration(Reference Jensen, Koh-Banerjee and Franz310). Data from the Nurses’ Health Study showed lower CRP and sTNFR2 concentrations in diabetic women with a moderate intake of whole grain compared with those with a low intake(Reference Qi, van Dam and Liu314). In another study with diabetic males (n 780; from the Health Professionals’ Follow-Up Study), Qi et al. (Reference Qi, Rimm and Liu315) showed that cereal fibre was positively associated with adiponectin concentration after controlling for a number of confounding factors. A higher intake of whole grain (low 0·8 servings/d, high 2·1 servings/d) within a Mediterranean diet in diabetic women was associated with increased adiponectin concentration(Reference Mantzoros, Williams and Manson312). Data from the Multi-Ethnic Study of Atherosclerosis (MESA) showed a higher whole grain intake (Q1 0·02 servings/d, Q5 1·39 servings/d) to be associated with lower CRP concentration in elderly subjects(Reference Lutsey, Jacobs and Kori311).
Table 3 Observational studies on the association between whole grain intake and markers of low-grade inflammation

M, male; F, female; ↓ , decreased; CRP, C-reactive protein; = , no effect on; sTNFR, soluble TNF receptors; sICAM, soluble intercellular adhesion molecule; sE-selectin, soluble E-selectin; ↑ , increased; PAI-1, plasminogen activator inhibitor 1.
Evidence from intervention studies (Table 4)(Reference Esposito, Pontillo and Di Palo111, Reference Andersson, Tengblad and Karlstrom316–Reference Tighe, Duthie and Vaughan320) includes a study whereby overweight and obese subjects consumed a hypoenergetic diet with or without whole-grain foods. CRP concentration decreased by 38 % (CRP at baseline 5·9–6·0 mg/l) in the whole-grain group independent of the observed weight loss(Reference Katcher, Legro and Kunselman319). However, plasma concentrations of IL-6 and TNF-α and PAI-1 activity did not change within the 12-week-study period. Based on the outcome of this study, the authors concluded that the changes seen in CRP concentration with a whole-grain diet were not part of a systemic anti-inflammatory effect(Reference Katcher, Legro and Kunselman319). In a study from Sweden with a similar design but only a 6-week intervention (whole-grain v. refined-grain products), the whole grain intake was not associated with changes in the concentrations of CRP and IL-6 or in PAI-1 activity of moderately overweight subjects(Reference Andersson, Tengblad and Karlstrom316). Baseline CRP concentration (2·03–2·86 mg/l) and BMI were much lower than in the previous study, which may explain the different outcomes between these studies. Recent intervention trials with whole grain (60–120 g/d) or wholemeal intake (30–40 g/d) and comparable CRP baseline concentrations also did not observe any changes in plasma markers of inflammation(Reference Brownlee, Moore and Chatfield317, Reference Tighe, Duthie and Vaughan320). In the study by Brownlee et al. (Reference Brownlee, Moore and Chatfield317), whole-grain products did not substitute refined-grain products but were consumed in addition to the refined products. Bioprocessing of whole wheat affected the anti-inflammatory potential in a human intervention study when compared with unprocessed whole wheat(Reference Mateo, Aura and Selinheimo321). Bioprocessing modulated the ratio of pro-inflammatory:anti-inflammatory cytokines during 24 h after the intake of 300 g whole wheat bread. This study suggests that subtle changes in the processing of whole grains may be relevant for their anti-inflammatory activity, which could in part explain the contradictory findings of the human intervention studies.
Table 4 Intervention studies investigating the effect of whole grain intake on markers of low-grade inflammation

M, male; F, female; ↓ , decreased; = , no effect on; CRP, C-reactive protein; PAI-1, plasminogen activator inhibitor-1; sVCAM-1, soluble vascular cell adhesion molecule-1; sICAM-1, soluble intercellular adhesion molecule-1; sE-selectin, soluble E-selectin.
Replacing a refined-wheat flour pizza by a similar pizza prepared from whole-wheat flour resulted in a decreased postprandial concentration of the pro-inflammatory cytokine IL-18 in both non-diabetic and diabetic subjects(Reference Esposito, Nappo and Giugliano318).
In summary, whole grain intake appears to inversely associate with markers of low-grade inflammation. Processing status of whole-grain products should be more precisely defined in future studies. Potential mechanisms still have to be elucidated, as well as the active constituents which may include dietary fibre, minerals, vitamins and phytochemicals such as lignans and phenolic acids.
Vegetables and fruits
A number of cross-sectional studies have investigated the association between vegetable and fruit intake and biomarkers of inflammation (Table 5)(Reference Salas-Salvado, Garcia-Arellano and Estruch288, Reference Bhupathiraju and Tucker322–Reference Yannakoulia, Yiannakouris and Melistas330). Study participants included healthy, normal-weight adults as well as overweight/obese adults with associated diseases. Each study applied different criteria to stratify the intake of vegetables and fruits which makes it difficult to compare the outcomes. Of the ten cross-sectional studies, seven reported an inverse association between a high intake of vegetables and fruits, either in combination or alone, and blood CRP concentrations(Reference Bhupathiraju and Tucker322–Reference Gao, Bermudez and Tucker325, Reference Holt, Steffen and Moran327–Reference Wannamethee, Lowe and Rumley329). In one study, no association with CRP was observed(Reference Salas-Salvado, Garcia-Arellano and Estruch288), while in a second study, an inverse association was seen in men but not in women(Reference Holt, Steffen and Moran327). For the other biomarkers reported, the outcome is less consistent. A recent study reported that a high intake of vegetables and fruits was associated with a lower peripheral blood mononuclear cell gene expression for several pro-inflammatory cytokines and adhesion molecules(Reference Hermsdorff, Zulet and Puchau326). An important observation is that besides the quantity of total intake of vegetables and fruits, the variety consumed in a given time has a significant impact on these biomarkers. A high number of varieties of vegetables and fruits were inversely correlated with blood CRP levels(Reference Bhupathiraju and Tucker322). This suggests that plant-specific constituents of vegetables and fruits such as the phytochemicals may contribute to the anti-inflammatory activities.
Table 5 Observational studies on the association between vegetable and fruit intake and markers of low-grade inflammation

M, male; F, female; ↓ , decreased; CRP, C-reactive protein; = , no effect on; sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular cell adhesion molecule-1; ↑ , increased; IL-1ra, IL-1 receptor antagonist.
A number of intervention studies have investigated the impact of vegetables and fruits in total or of specific varieties on biomarkers of inflammation (Table 6)(Reference Basu, Du and Leyva331–Reference Zern, Wood and Greene346). Of the four studies focusing on vegetables and fruits as a food group, three reported a reduction in blood concentrations of different biomarkers of inflammation(Reference Esposito, Nappo and Giugliano318, Reference Sanchez-Moreno, Cano and de342, Reference Watzl, Kulling and Moseneder345), while one study did not find any significant effect(Reference Freese, Vaarala and Turpeinen336). In twelve studies with a specific focus on a single variety of vegetable or fruit, inconsistent results were reported. Most studies have used fruits or fruit extracts high in polyphenols. Results from such studies suggest an anti-inflammatory effect; however, mostly, only a single biomarker was affected, and never the complete set of inflammation biomarkers investigated(Reference Basu, Du and Leyva331–Reference Dalgard, Nielsen and Morrow334, Reference Karlsen, Retterstol and Laake337–Reference Larmo, Alin and Salminen340, Reference Zern, Wood and Greene346). In conclusion, current evidence for specific effects of single vegetable and fruit varieties is not convincing, while a high overall intake of vegetables and fruits seems to be associated with a lower state of inflammation.
Table 6 Intervention studies investigating the effect of vegetable and fruit intake on markers of low-grade inflammation

M, male; F, female; ↓ , decreased; PAI-1, plasminogen activator inhibitor-1; = , no effect on; CRP, C-reactive protein; MCP-1, monocyte chemoattractant protein-1; sICAM-1, soluble intercellular adhesion molecule-1; sP-selectin, soluble P-selectin; sVCAM-1, soluble vascular cell adhesion molecule-1; sE-selectin, soluble E-selectin; RANTES, regulated on activation, normal T expressed and secreted; IFN, interferon; sTNFR, soluble TNF receptor; MIG, monokine-induced by IFN-γ; WBC, white blood cell; IL-1ra, IL-1 receptor antagonist; ↑ , increased.
Soya
Several randomised controlled intervention trials have shown that supplementation with different quantities of soya protein did not affect markers of inflammation (CRP, IL-6, IL-18, sICAM-1, sVCAM-1 and sE-selectin) (Table 7)(Reference Watzl, Kulling and Moseneder345, Reference Azadbakht, Kimiagar and Mehrabi347–Reference Zemel, Sun and Sobhani364). Variations in soya protein isoflavone contents did not modulate the outcome in these studies. In one study, reduced plasma CRP, TNF-α and IL-18 concentrations were reported in postmenopausal Iranian women with the metabolic syndrome consuming soya nuts but not soya protein(Reference Azadbakht, Kimiagar and Mehrabi347). The major difference between soya protein and soya nuts (comparable contents of isoflavones) was a much higher content of PUFA in the soya nuts, suggesting that rather than soya-specific constituents, the increased intake of PUFA may be responsible for the reduction in biomarkers of inflammation(Reference Azadbakht, Kimiagar and Mehrabi347). The same group investigated in diabetic patients with nephropathy the anti-inflammatory effect of textured soya protein compared with animal protein after 4 years of intake. Consumption of soya protein significantly lowered plasma concentrations of CRP(Reference Azadbakht, Atabak and Esmaillzadeh348). In another study investigating soya nuts in normotensive and hypertensive postmenopausal women, no effect on CRP, IL-6, sICAM-1 or matrix metalloproteinase-9 concentrations was observed compared with the control diet(Reference Nasca, Zhou and Welty361). However, in hypertensive women, levels of sVCAM-1 were significantly lower after soya nut consumption(Reference Nasca, Zhou and Welty361). Again, the increased PUFA intake may have caused this effect. Long-term soya exposure did not affect CRP, IL-6, leptin and adiponectin concentrations in postmenopausal women(Reference Maskarinec, Steude and Franke357), or CRP, IL-6, leptin, adiponectin, MCP-1 or MIP-1β concentrations in men(Reference Maskarinec, Oum and Chaptman365). Short-term soya intervention in postmenopausal women with or without exercise-induced inflammation had no effect on IL-1β, IL-6 or TNF-α concentrations(Reference Beavers, Serra and Beavers349, Reference Beavers, Serra and Beavers350). Among men with prostate cancer undergoing androgen deprivation therapy, soya intervention had no effect on the concentrations of several inflammatory markers(Reference Napora, Short and Muller366). Overall, these data suggest that soya protein does not affect markers of inflammation, that soyabean processing may affect the anti-inflammatory potential of soyabean constituents and that the health status of the study subjects might determine the anti-inflammatory efficacy of specific foods.
Table 7 Intervention studies investigating the effect of soya intake on markers of low-grade inflammation

F, female; = , no effect on; sIL-2R, soluble IL-2 receptor; sE-selectin, soluble E-selectin; sP-selectin, soluble P-selectin; sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular cell adhesion molecule-1; CRP, C-reactive protein; M, male; ↓ , decreased; ↑ , increased; MCP, monocyte chemoattractant protein; S-hs CRP, serum high-sensitivity C-reactive protein; DASH, dietary approaches to stop hypertension.
Nuts
To date, only few studies have investigated the effect of nuts on inflammatory markers. The MESA reported that a high intake of nuts and seeds ( ≥ 5 times/week) compared with a low intake (rarely or no consumption) was associated with lower plasma concentrations of CRP, IL-6 and fibrinogen(Reference Jiang, Jacobs and Mayer-Davis367). In contrast, data from the Nurses’ Health Study suggest that nut intake is not associated with inflammatory markers including sTNFR2, CRP, fibrinogen, sICAM-1 and sE-selectin(Reference Li, Brennan and Wedick368). An 8-week intervention with walnuts or cashews (63–108 g/d) in subjects with the metabolic syndrome observed no effect on serum CRP concentrations(Reference Mukuddem-Petersen, Stonehouse and Jerling360). A pistachio intervention (60–100 g/d providing about 20 % of total energy) for 4 weeks in healthy young subjects had no effect on plasma CRP or TNF-α concentrations, while the concentration of IL-6 was significantly reduced(Reference Sari, Baltaci and Bagci369). A recent intervention study with two doses of almonds (10 and 20 % of total energy) in healthy adults reported reduced serum concentrations of sE-selectin and CRP, but IL-6 and fibrinogen were not affected(Reference Rajaram, Connell and Sabate370). No clear dose–response relationships were observed. In contrast, an earlier study in hyperlipidaemic subjects providing two doses of almonds did not observe an effect on CRP concentrations(Reference Jenkins, Kendall and Jackson356). A systematic review on walnut consumption and inflammatory markers also reported inconsistent results for plasma CRP, while walnuts added to a Mediterranean diet resulted in significantly lower sVCAM-1 concentrations(Reference Banel and Hu371). This suggests that rather than a general anti-inflammatory effect, walnuts may exert anti-inflammatory effects primarily in the endothelium. Major contributors to any anti-inflammatory activity of nuts are likely to include PUFA, Mg and phytochemicals including ellagic acid(Reference Ros372).
Fish
Increased frequency of fish consumption was associated with lower CRP and IL-6 concentrations in a cohort of 727 adults in the USA(Reference Lopez-Garcia, Schulze and Manson373). sICAM-1 and sE-selectin concentrations also decreased but the effect of fish consumption was weaker than what was observed for CRP and IL-6. sTNFR2 concentration was not associated with fish consumption. A study in 379 adults in Denmark reported a lack of association between fish consumption and CRP concentration, even though the subjects displayed a wide range of intakes including very high intakes (e.g. over 30 % of subjects ate fish more than once per d)(Reference Madsen, Skou and Hansen374). Data from 5037 adults in the NHANES showed no association of fish consumption with CRP concentration after adjusting for a range of confounders(Reference King, Egan and Geesey375). In this study, 25 % of subjects consumed fish more than 3 times per month. Most recently, fish consumption among a cohort of 4077 Australian adults was reported not to be associated with CRP concentration before or after adjustment for confounders(Reference Hickling, Hung and Knuiman376). In contrast to these findings, a study in 3102 Greek adults reported that fish consumption was ‘dose-dependently’ associated with lower CRP, IL-6, TNF-α and SAA concentrations and white blood cell counts; individuals consuming >300 g fish/week (n 259; 9 %) had significantly lower concentrations of CRP ( − 33 %), IL-6 ( − 33 %), TNF-α ( − 21 %), SAA ( − 28 %) and leucocytes ( − 4 %) than seen in individuals not consuming fish (n 319; 11 %)(Reference Zampelas, Panagiotakos and Pitsavos377). It is possible that the observations of Zampelas et al. may be due to greater fish consumption than in the other populations studied and/or to the type of fish consumed. No studies have discriminated between consumption of lean v. fatty fish. A 4-week intervention study with herring (150 g/d, 5 d/week) in overweight and obese adults in Sweden (n 13) reported a trend towards lower CRP concentration compared with the concentration seen when the subjects consumed a reference diet, but the 30 % reduction in CRP was not significant(Reference Lindqvist, Langkilde and Undeland378). The lack of statistical significance of the effect seen may be due to the small sample size.
Tea
A cross-sectional study from Japan reported no relationship between green tea consumption and the concentrations of several inflammatory markers(Reference Maki, Pham and Yoshida379), while a cross-sectional study from Belgium detected significantly lower CRP and SAA concentrations in regular tea drinkers after multivariate analysis(Reference De Bacquer, Clays and Delanghe380). The effect of tea consumption (green or black tea) has been investigated in several small intervention studies usually with a fairly short duration (Table 8)(Reference Basu, Du and Sanchez381–Reference Widlansky, Duffy and Hamburg389). Most studies indicate that tea consumption has no significant effect on the markers of inflammation reported(Reference de Maat, Pijl and Kluft382–Reference Ryu, Lee and Lee386, Reference Sung, Min and Lee388, Reference Widlansky, Duffy and Hamburg389). However, a longer supplementation period of 6 weeks with black tea in healthy non-smoking men reduced plasma CRP concentrations significantly(Reference De Bacquer, Clays and Delanghe380, Reference Steptoe, Gibson and Vuononvirta387). Overall, there are no consistent data that tea consumption (black or green) has a beneficial effect on inflammatory status, but studies of longer duration than those currently reported need to be performed.
Table 8 Intervention studies investigating the effect of black or green tea intake on markers of low-grade inflammation

M, male; F, female; = , no effect on; CRP, C-reactive protein; sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular cell adhesion molecule-1; ↓ , decreased; SAA, serum amyloid A.
The modest or even absent effects of tea on inflammatory and oxidative stress markers in vivo are surprising in view of the potent inhibitory effects of tea components such as catechins on the expression of pro-inflammatory mediators in vitro (Reference Khan, Afaq and Saleem390–Reference Syed, Afaq and Kweon392). This may be due to the fact that the majority of tea catechins undergo methylation, glucuronidation and sulfation during uptake which may limit bioavailability(Reference Feng393). Furthermore, studies in vitro have often used catechin concentrations >10 μm, whereas plasma concentrations of catechins after ingestion of tea rarely exceed 1 μm(Reference Renouf, Guy and Marmet394).
Coffee
Habitual coffee consumption was analysed for association with markers of low-grade inflammation in cross-sectional epidemiological studies which yield conflicting results (Table 9)(Reference Schulze, Hoffmann and Manson304, Reference Hamer, Williams and Vuononvirta395–Reference Zampelas, Panagiotakos and Pitsavos398). Consumption of decaffeinated coffee was not associated with changes in systemic levels of soluble adhesion molecules, but CRP concentrations were lower in decaffeinated coffee drinkers in one of the two studies(Reference Lopez-Garcia, van Dam and Qi396). The contradictory findings from observational studies may reflect the fact that coffee contains a mixture of bioactives with divergent effects on physiology(Reference Rodrigues and Klein399–Reference Battram, Arthur and Weekes403). A recent intervention study providing two doses of coffee (4 and 8 cups of filtered coffee/d) in the same individuals confirmed in part the observations from the cross-sectional studies. While IL-18 (decreased) and adiponectin (increased) were significantly modulated by coffee consumption, concentrations of CRP, leptin, SAA, IL-6, MIF and IL-1ra were not affected(Reference Kempf, Herder and Erlund404). No dose–response relationship was seen in this study. Plasma caffeine concentrations were positively associated with plasma adiponectin concentrations(Reference Kempf, Herder and Erlund404). Taken together, the available data do not allow a firm conclusion as to whether coffee consumption modulates low-grade inflammation.
Table 9 Observational studies on the association between coffee intake and markers of low-grade inflammation

M, male; F, female; ↑ , increased; CRP, C-reactive protein; SAA, serum amyloid A; = , no effect on; sE-selectin, soluble E-selectin; sICAM-1, soluble intercellular adhesion molecule-1; sTNFR, soluble TNF receptor; ↓ , decreased; sVCAM-1, soluble vascular cell adhesion molecule-1.
Cocoa
Cocoa has a high content of monomeric (epicatechin and catechin) and oligomeric (procyanidin) flavanols(Reference Adamson, Lazarus and Mitchell405–Reference Natsume, Osakabe and Yamagishi407). These latter polymeric fractions are present in higher concentrations/amounts in cocoa compared with other flavanol-rich foods such as red wine or green tea(Reference Lazarus, Adamson and Hammerstone408, Reference Lazarus, Hammerstone and Schmitz409). Thus, certain cocoa-based products are rich in flavanols(Reference Vinson, Jang and Dabbagh410, Reference Hammerstone, Lazarus and Schmitz411), some of which have been found in model systems to possess potential anti-inflammatory activities. However, the effect of cocoa flavanols and their related procyanidins appears to be related to the degree of polymerisation, and opposing effects on inflammatory cytokine production in vitro of low- and higher-degree of polymerisation flavanols have been reported(Reference Mao, Van de Water and Keen412, Reference Mao, Powell and Van de Water413). Because of the focus on in vitro exploration of the effects of cocoa flavanols, there is a need for well-designed human studies, using cocoa properly characterised in terms of flavanol content. In an Italian study of over 10 000 people, of whom half were free of any chronic disease, 1317 people reported having eaten any chocolate during the past year; 824 ate chocolate regularly in the form of dark chocolate only(Reference di Giuseppe, Di Castelnuovo and Centritto414). Regular consumption of small doses of dark chocolate seemed to decrease inflammation: after adjustment for multiple confounders, dark chocolate consumption was inversely associated with CRP concentration. Serum CRP concentrations were 1·32 (1·26–1·39 mg/l) in chocolate non-consumers and 1·10 (1·03–1·17 mg/l) in consumers. A J-shaped relationship between dark chocolate consumption and serum CRP was observed; consumers of up to 1 serving (20 g) of dark chocolate every 3 d had significantly lower serum CRP concentrations than non-consumers or those consuming more than 20 g chocolate per d. In a small uncontrolled intervention study, healthy subjects (n 25) consumed dark chocolate (36·9 g/d) and a cocoa powder drink (30·95 g powder/d) for 6 weeks; there was no change in concentrations of CRP, TNF-α, IL-1β, IL-6 or soluble P-selectin (sP-selectin)(Reference Mathur, Devaraj and Grundy415). Another small (n 28), uncontrolled intervention study with dark chocolate for 1 week found a reduction in CRP concentration in women but not in men(Reference Hamed, Gambert and Bliden416). Most recently, Monagas et al. (Reference Monagas, Khan and Andres-Lacueva417) reported the effect of a 4-week randomised cross-over trial of 40 g cocoa powder in skimmed milk daily v. skimmed milk in forty-two older subjects: serum concentrations of sICAM-1 and sP-selectin were lower after the cocoa powder intervention. Thus, there is some evidence that cocoa and cocoa-rich foods reduce low-grade inflammation.
Alcohol
The regular consumption of alcohol-containing beverages such as wine or beer has been reported to be inversely associated with several markers of low-grade inflammation, in a dose-dependent manner: i.e. moderate daily intake (1–2 drinks/d) was often found to be associated with decreased concentrations of inflammatory markers, although this was not the case in all studies (Table 10) (Reference Albert, Glynn and Ridker418–Reference Wannamethee, Lowe and Shaper432).
Table 10 Observational studies on the association between alcohol intake and markers of low-grade inflammation

M, male; F, female; U, U-shaped relationship; CRP, C-reactive protein; ↑ , increased; = , no effect on; ↓ , decreased; sICAM-1, soluble intercellular adhesion molecule-1; SAA, serum amyloid A; iU, inverse U-shaped relationship; sTNFR, soluble TNF receptor; sVCAM-1, soluble vascular cell adhesion molecule-1; PAI-1, plasminogen activator inhibitor 1.
The short-term effect of alcohol intake on markers of low-grade inflammation has been studied in intervention trials which either determined acute effects during the next hours following ingestion or changes compared with baseline after 2–4 weeks of daily alcohol consumption (Table 11)(Reference Beulens, de Zoete and Kok433–Reference Williams, Sutherland and Whelan448). Although most studies were of small size, findings were quite consistent in that there was little change in low-grade inflammation in the hours following alcohol intake. In one study, a decrease in NF-κB activation was reported after consuming a fat-rich meal when red wine was also consumed compared with consuming the fat-rich meal(Reference Blanco-Colio, Valderrama and Alvarez-Sala438). In general, findings are fairly similar for trials with wine, beer, vodka or ethanol (see Table 11). After 2–4 weeks of daily intake of alcoholic beverages, changes in low-grade inflammation were noted in five out of eight trials, with decreases in concentrations of CRP, cytokines and soluble adhesion molecules, and an increased concentration of adiponectin (see Table 11). These effects were seen in trials with white or red wine, or beer. The three ‘negative’ trials(Reference Beulens, de Zoete and Kok433–Reference Watzl, Bub and Pretzer436) used red wine or beer. Some variation of outcome between trials is to be expected, notably because the lifestyle and characteristics of participants will not be identical between trials, in particular with regard to the local diet and the amount of daily physical activity. Nevertheless, when taken together, these studies suggest some beneficial long-term effects of moderate daily consumption of wine or beer on low-grade inflammation. It is not clear whether such effects are to due to the alcohol content of the beverages tested, or to other components, such as phenolic compounds in these fermented beverages.
Table 11 Intervention studies investigating the effect of alcohol intake on markers of low-grade inflammation

M, male; F, female; ↓ , decreased; = , no effect on; sP-selectin, soluble P-selectin; ↑ , increased; CRP, C-reactive protein; sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular cell adhesion molecule-1; TGF, transforming growth factor; MCP-1, monocyte chemoattractant protein-1; sCD40L, sCD40 ligand.
Process-related compounds: advanced glycation end products and advanced lipoperoxidation end products
Introduction
Humans have been using fire to cook food for thousands of years. They used boiling, baking, broiling, roasting, grilling or frying to make food more hygienic, digestible, nutritive and durable, and particularly to improve its flavour, aroma and texture. Despite man's long history of exposure to heated food, the demonstration that the Maillard reaction also occurs and is associated with ageing and age-related conditions, such as diabetic complications, atherosclerosis, Alzheimer's disease and hypertension, has raised the question whether dietary Maillard reaction products (MRP) might represent a risk to human health. A plausible mechanism for adverse health effects of dietary MRP could be their potential promotion of a state of low-grade inflammation. In fact, consumption of a Western diet rich in these products was found to correlate with impaired glucose metabolism, insulin resistance and increased risk of cardiovascular and renal disease associated with the metabolic syndrome and related conditions(Reference Vlassara449, Reference Vlassara and Striker450). However, although excellent work has been published in the past few years, it is still unresolved whether MRP are causally involved in the aetiology of these conditions(Reference Ames451–Reference Sebekova and Somoza453). In particular, whether the potential harm of a Western diet is mainly due to the increased intake of Maillard compounds produced by heating of carbohydrate- and/or lipid-rich food or rather to the excess intake of energy, refined sugars and saturated fats, together with the lower intake of fresh fruits and vegetables, can be debated. Alternatively, other factors generated by thermal treatment of food or the thermal destruction of vitamins, polyphenols and antioxidants during food processing could also contribute to adverse health effects.
Advanced glycation end products and advanced lipoxidation end products
Advanced glycation end products (AGE) are a family of heterogeneous, partly uncharacterised compounds that encompass both pre-melanoidins and melanoidins. Pre-melanoidins are not coloured and in general exhibit no fluorescence, whereas melanoidins are yellow to brown and are often fluorescent. The pre-melanoidins include uncoloured non-cross-linking AGE such as pyrraline, carboxymethyllysine and carboxyethyllysine(Reference Henle454, Reference Monnier455). The melanoidins include all the fluorescent cross-linked AGE formed in the ‘non-enzymatic browning reaction’, such as pentosidine, and crossline, but also non-fluorescent cross-linked AGE such as glyoxal lysine dimer, methylglyoxal lysine dimer and the alkyl formyl glycosyl pyrroles, formed by the reaction between two sugar molecules with a single lysine residue, and arginine-lysine imidazole, believed to be an intermolecular cross-link. In addition, some compounds that are not AGE in the strict sense are sometimes also considered as AGE. These include the products of the Amadori rearrangement such as fructoselysine (formed with glucose) and the unstable, reactive dicarbonyl intermediates formed by the breakdown of sugars or Amadori intermediates, which may be included in the pre-melanoidins. AGE accumulate in the circulation under several physiological and pathological conditions, including diabetes and related disorders. In addition, circulating AGE accumulate when renal function is impaired(Reference Koschinsky, He and Mitsuhashi456–Reference Uribarri, Peppa and Cai459). AGE are formed within the body (endogenous AGE) due to at least three mechanisms(Reference Baynes and Thorpe460):
(1) Enhanced carbohydrate (and/or lipid) substrate availability, such as in hyperglycaemia (and/or hyperlipidaemia), which favours the nucleophilic addition of a carbonyl group from a reducing sugar to a free amino group of a protein (protein glycation) to form a reversible Schiff's base, which then rearranges to the more stable ketoamine or Amadori product and subsequently, through dicarbonyl intermediates such as 3-deoxyglucosones, to the irreversible AGE.
(2) Increased oxidative metabolism, such as in the presence of transition metals and oxidative stress, which cause auto-oxidation of glucose (auto-oxidative glycation) or Amadori products (glycoxidation) via formation of dicarbonyl compounds, such as glyoxal.
(3) Increased non-oxidative metabolism, with accumulation of reducing sugars other than glucose, such as in the case of increased glucose flux through the glycolysis and polyol pathway (intracellular glycation) and/or thiamin deficiency, which result in the formation of the AGE precursor methylglyoxal.
AGE may also be derived from food(Reference Henle454, Reference Goldberg, Cai and Peppa461) or tobacco(Reference Cerami, Founds and Nicholl462). AGE are formed during cooking and food processing procedures, and could accumulate in the body after intestinal absorption. It has been suggested from experimental animal models and studies in both healthy and type 2 diabetic subjects that dietary AGE represent a significant source of circulating and tissue AGE, although the absorption of individual AGE from food is largely unknown(Reference Erbersdobler and Faist463). Little is known about possible tissue deposition of food-derived AGE but in healthy animals, it has been shown that AGE are rapidly and completely excreted(Reference Bergmann, Helling and Heichert464, Reference Finot and Magnenat465). Finally, accumulation of both endogenous and exogenous AGE may be favoured by reduced kidney clearance as in the case of renal failure(Reference Ahmed466) and by impaired detoxification caused by the utilisation of cofactors of detoxifying enzymes.
The so-called advanced lipoxidation end products (ALE) are similar to AGE but, rather than originating from sugars and sugar breakdown products, ALE are derived from lipid oxidation(Reference Hidalgo and Zamora467). Metal-catalysed oxidation of unsaturated lipids such as PUFA and cholesterol results in the formation of lipid hydroperoxides and oxycholesterols, respectively. Lipid hydroperoxides, in the presence of metal ions or at high temperature, form epoxyhydroperoxides, ketohydroperoxides and cyclic peroxides, which decompose to low-molecular-weight breakdown products such as aldehydes, ketones or alcohols, or condense to polymers. Malondialdehyde and 4-hydroxy-2-nonenal are the main aldehydes generated during lipid peroxidation of n-6 PUFA(Reference Esterbauer, Schaur and Zollner468), whereas 4-hydroxy-2-hexanal is the predominant compound derived from the oxidation of n-3 PUFA(Reference Uchida469). Reactive lipid peroxidation products can form ALE by reacting with amino groups of proteins to generate labile or stable adducts or cross-links in protein, some of which may be coloured or fluorescent. Some reaction products, such as carboxymethyllysine, can originate from both sugars and lipid oxidation products(Reference Fu, Requena and Jenkins470), and have been called ‘either advanced glycation and lipoxidation end products’(Reference Ahmed466).
Advanced glycation end products/advanced lipoxidation end products receptors
The putative deleterious effects of AGE may be attributed to direct physico-chemical effects such as modification of extracellular matrix proteins, resulting in cross-links, leading to increased vascular stiffness or modification of proteins resulting in altered function, as well as via binding to a variety of cell-surface receptors(Reference Brownlee471). AGE receptors have a dual function and are involved in both AGE removal and AGE-induced cell activation(Reference Yan, Barile and D'Agati140). Cell activation can occur via receptor-mediated generation of ROS through both mitochondrial and cytosolic pathways involving the electron transport chain and NAD(P)H oxidase, respectively(Reference Basta, Lazzerini and Del472, Reference Jay, Hitomi and Griendling473). ROS can trigger pro-inflammatory signalling pathways causing MAPK-dependent activation of transcription factors such as NF-κB(Reference Bierhaus, Humpert and Morcos474–Reference Lander, Tauras and Ogiste476) and consequent modulation of the gene expression of several pro-inflammatory cytokines(Reference Bierhaus, Humpert and Morcos474, Reference Bierhaus, Rudofsky and Humpert475, Reference Ramasamy, Yan and Schmidt477).
Several AGE-binding proteins have been identified, including the 42–45 kD receptor for AGE (RAGE), a member of the Ig superfamily(Reference Stern, Yan and Yan478), the 60 kD protein a 48-kDa member of the oligosaccharyltransferase complex (OST-48) or AGE-receptor (AGE-R)1, the 90 kD protein 80K-H or AGE-R2(Reference Li, Mitsuhashi and Wojciechowicz479), and the 32 kD protein galectin-3 or AGE-R3(Reference Vlassara, Li and Imani480). This redundancy could imply binding and/or functional specificity among AGE receptors; alternatively, not all these receptors might be relevant for AGE binding in vivo (Reference Yan, Barile and D'Agati140, Reference Heizmann481). At present, RAGE seems to be the only receptor involved in cell activation(Reference Yan, Barile and D'Agati140). On the other hand, AGE-R1, AGE-R2 and AGE-R3 seem to behave as an AGE-receptor complex rather than individual receptor molecules and exert a predominant scavenging function(Reference Cai, He and Zhu482, Reference Iacobini, Amadio and Oddi483).
In addition to the classical AGE receptors, AGE are cleared also by scavenger receptors, which share with AGE receptors the ability to bind modified lipoproteins such as oxidised LDL(Reference Horiuchi, Sakamoto and Sakai484). The scavenger receptor family can be broadly classified into eight classes (A–H), which are expressed on macrophages but also on other cells, including endothelial, mesangial and vascular smooth muscle cells(Reference Greaves and Gordon485, Reference Murphy, Tedbury and Homer-Vanniasinkam486). Though the ability of AGE receptors and scavenger receptors to clear pro-inflammatory compounds such as AGE and oxidised LDL from the tissue or circulation would seem initially beneficial, the final net effect is dependent upon the balance between this scavenging function and several other functions of these receptors, including the clearance of other modified self and non-self ligands, downstream pathways with formation of cholesterol-laden cells, the initiation of pro-inflammatory signalling cascades, and the regulation of cellular lipid influx/efflux and synthesis/degradation(Reference Moore and Freeman487). However, it should be emphasised that the ligand affinity of AGE proteins to scavenger receptors is dependent on their modification, with mildly modified AGE not showing significant affinity(Reference Nagai, Mera and Nakajou488).
Among the AGE receptors, RAGE was shown to be implicated in atherogenesis, based on the observations that soluble RAGE, which is able to bind circulating AGE and other RAGE ligands, and therefore to prevent their pro-inflammatory effects, inhibited the development of atherosclerosis in diabetic apoE− / − mice(Reference Park, Raman and Lee489), arrested its progression when treatment was started after establishment of lesions(Reference Bucciarelli, Wendt and Qu490) and prevented experimental diabetic nephropathy(Reference Flyvbjerg, Denner and Schrijvers491), which was conversely accelerated by RAGE overexpression(Reference Yamamoto, Kato and Doi492). In addition, RAGE ablation prevented diabetic nephropathy(Reference Wendt, Tanji and Guo493) and neuropathy(Reference Bierhaus, Haslbeck and Humpert494, Reference Haslbeck, Schleicher and Bierhaus495). Thus, the cell-surface RAGE appears to be involved in inducing pro-inflammatory signalling while the soluble form of RAGE appears to prevent this, by sequestering ligands away from the cell-surface RAGE. Conversely, galectin-3 was proposed to exert a prevailing protective role as indicated by reports that galectin-3 ablation resulted in (1) the acceleration of glomerulopathy induced by diabetes(Reference Pugliese, Pricci and Iacobini496), (2) increased AGE levels(Reference Iacobini, Menini and Oddi497) and (3) accelerated ageing(Reference Iacobini, Oddi and Menini498). In the vessel wall, galectin-3 was induced in proliferating vascular smooth muscle cells and particularly in foam cells from arteries of experimental animal models of atherosclerosis and human patients with advanced atherosclerotic lesions(Reference Arar, Gaudin and Capron499, Reference Nachtigal, Al-Assaad and Mayer500), and recent studies have shown that galectin-3 ablation accelerates lipid-induced atherogenesis(Reference Iacobini, Menini and Ricci501). Also AGE-R1 seems to play a protective role, as indicated by resistance to oxidant and inflammatory injury in mesangial cells overexpressing this receptor(Reference Cai, He and Zhu482, Reference Lu, He and Cai502). This was supported by the association of low AGE-R1 expression with susceptibility to diabetic nephropathy in non-obese diabetic mice(Reference He, Zheng and Stitt503) and the presence of diabetic complications in type 1 diabetic patients(Reference He, Koschinsky and Buenting504). Finally, the participation of class A and B scavenger receptors in the process of atherogenesis has been clearly established by a large body of experimental studies, with scavenger receptor AI (SR-AI) and II and CD36 showing a predominant pro-atherogenic role and scavenger receptor BI (SR-BI) exerting a protective function towards lesion development(Reference Moore and Freeman487). Thus, the net effect of the interaction of AGE with AGE-binding receptors that can result in either clearance or cell activation would be determined by the relative expression of these different receptors on the respective cells.
Significance of dietary advanced glycation end products/advanced lipoxidation end products
The nutritional properties of heated food were initially questioned because of the reduced availability of lysine due to the formation of Amadori-modified lysine(Reference Finot, Bujard and Mottu505). However, cooking was also shown to increase the bioavailability of some nutrients, especially proteins derived from some plant sources(Reference Ames451). Subsequently, the concern was raised that AGE (and ALE) formed during the Maillard reaction might exert adverse health effects in cells, tissues and organs. As a potential mechanism, a role of AGE/ALE binding to RAGE followed by triggering of RAGE-dependent downstream signalling events leading to transcription of pro-inflammatory genes has been postulated(Reference Baynes and Thorpe460, Reference Vlassara and Palace506). This view is supported by a number of studies conducted in both experimental animal models and human subjects, showing that ingestion of diets containing high AGE levels results in increased levels of circulating AGE (see below). In addition, these high-AGE diets, generated by more severe thermal treatment than the control diets, also resulted in a series of metabolic and micro/macrovascular abnormalities related to diabetes and its long-term complications (see below). However, despite this body of experimental data, a cause–effect relationship between dietary AGE and development and progression of disease conditions related to Western-type lifestyle and dietary habits has not yet been conclusively demonstrated, and a number of issues remain to be solved.
Evidence supporting a potential harmful effect of dietary advanced glycation end products/advanced lipoxidation end products
Animal studies
In spontaneously diabetic db/db mice(Reference Hofmann, Dong and Li507) and in normal mice fed high-fat diets(Reference Sandu, Song and Cai508), feeding of a low-AGE diet reduced serum AGE levels and improved insulin sensitivity, compared with a high-AGE diet. In addition, in non-obese diabetic mice, a fetal or neonatal low-AGE environment prevented autoimmune diabetes, possibly by reducing antigenic stimulus for T-cell-mediated injury or by attenuating β-cell damage(Reference Peppa, He and Hattori509). In both type 1 and 2 diabetes models (i.e. non-obese diabetic and db/db mice, respectively) low dietary AGE content provided sustained protection towards the development of diabetic nephropathy(Reference Zheng, He and Cai510). Likewise, in genetically hypercholesterolaemic apoE− / − mice, low AGE content in the diet attenuated the development of atherosclerosis after induction of diabetes with streptozotocin(Reference Lin, Choudhury and Cai511) and neointimal formation after arterial injury(Reference Lin, Reis and Dore512). Finally, in db/db mice, wound healing was accelerated by a low-AGE diet, with predominant wound contraction, compared with animals on a high-AGE diet, which favoured the epithelialisation mode of wound closure(Reference Peppa, Brem and Ehrlich513).
Human studies
In healthy younger (aged < 45 years) and older (aged >60 years) human subjects, dietary AGE, but not energy intake, correlated with serum AGE levels and markers of oxidative stress (8-isoprostanes), which in turn correlated with serum CRP concentration and insulin resistance(Reference Negrean, Stirban and Stratmann514–Reference Vlassara, Cai and Crandall521) (Table 12). It is noteworthy that in this study, some healthy individuals showed AGE levels of similar magnitude as found in diabetic patients.
Table 12 Studies on the association between advanced glycation end products (AGE) intake and markers of low-grade inflammation

M, male; F, female; ↑ , increased; CRP, C-reactive protein; sVCAM-1, soluble vascular cell adhesion molecule-1; = , no effect on; PAI-1, plasminogen activator inhibitor-1; sICAM-1, soluble intercellular adhesion molecule-1; ↓ , decreased.
AGE-rich diets used for intervention studies in human subjects are usually generated by more severe thermal treatment of food, particularly by frying, broiling and baking, whereas AGE-poor food is prepared by steaming or simply without heating at all. Therefore, unless specified, these are the methods that were used for the preparation of test and control diets in the studies reviewed below. Taken together, these studies show that dietary AGE content correlates with circulating levels of AGE and with markers of inflammation, oxidative stress, endothelial dysfunction and renal function. One caveat that needs attention for future human studies is the significant contribution of smoking to the endogenous AGE load(Reference Cerami, Founds and Nicholl462, Reference Venkatesan, Hemalatha and Bobby522).
Diabetic subjects with and without nephropathy were given a single meal of egg-white, cooked with or without fructose, with the AGE-rich food containing about three times more AGE than a single meal of a regular diet(Reference Koschinsky, He and Mitsuhashi456). There was a significant correlation between dietary AGE content and serum AGE levels. Moreover, the serum AGE levels were directly related to the degree of renal dysfunction, as estimated by the increase in albuminuria and the decrease in creatinine clearance, and renal AGE excretion was inversely correlated with the degree of renal dysfunction. The relationship between dietary AGE intake, serum AGE levels and renal AGE clearance was confirmed in a cross-sectional study performed in long-term haemodialysis and peritoneal dialysis patients with or without diabetes. This study showed that circulating AGE correlated significantly with the AGE content of a regular diet, as estimated by means of 3 d dietary records and food questionnaires, as well as with blood urea N level(Reference Uribarri, Peppa and Cai459). Likewise, in non-diabetic patients on maintenance peritoneal dialysis randomly assigned to two groups consuming either a high- or low-AGE diet for 4 weeks, serum AGE levels correlated with AGE consumption as well as with blood urea N, serum creatinine, total protein, albumin and P, with AGE restriction profoundly reducing circulating AGE(Reference Uribarri, Peppa and Cai523). Finally, cross-sectional studies of healthy subjects and of patients undergoing haemodialysis showed a positive relationship between dietary AGE intake and plasma inflammatory markers(Reference Peppa, Uribarri and Cai515, Reference Uribarri, Cai and Peppa520).
A number of acute studies examining the effect of a single high-AGE meal on inflammatory markers have been conducted (see Table 12). Although these have been conducted in a variety of subject/patient groups, they show rather similar effects: typically, there is an elevation of various inflammatory markers (CRP, cytokines and soluble adhesion molecules) in the hours after consumption of a high-AGE meal, and this increase does not occur with a low-AGE meal (see Table 12). Furthermore, healthy and diabetic volunteers receiving a single dose of a high-AGE (caffeine-free) cola drink showed an increase in serum PAI-1(Reference Uribarri, Stirban and Sander519).
In patients on haemodialysis or peritoneal dialysis randomly assigned to a 4-week high- or low-AGE diet, circulating levels of AGE as well as markers of inflammation and endothelial dysfunction (which were markedly elevated at baseline, although not correlated with AGE) decreased significantly in response to AGE restriction, i.e. the low-AGE diet(Reference Peppa, Uribarri and Cai515). The findings of this latter study are in accordance with earlier data in diabetic subjects with normal renal function who showed a decrease in several inflammatory markers following 2 or 6 weeks of a low-AGE diet(Reference Vlassara, Cai and Crandall521).
Arguments against potentially harmful effects of dietary advanced glycation end products/advanced lipoxidation end products
In contrast to the aforementioned reports, circulating AGE levels could not be identified as an independent risk factor for cardiovascular or renal outcomes in diabetic subjects with diabetic nephropathy(Reference Busch, Franke and Wolf524), and circulating AGE levels were even inversely correlated with mortality in haemodialysis patients, the latter most probably due to a better nutritional status(Reference Schwedler, Metzger and Schinzel525). In addition, a recent study in obese children found lower circulating AGE levels compared with lean controls while inflammatory markers were significantly elevated in obese children(Reference Sebekova, Somoza and Jarcuskova517). Several issues concerning the methodology and interpretation of published data have been raised that may question the alleged adverse health effects of dietary AGE/ALE(Reference Ames451, Reference Baynes526, Reference Tessier and Birlouez-Aragon527).
A first issue is the large heterogeneity of AGE/ALE and their precursors, which would require examination of the effect of each of these compounds separately. In particular, the bioavailability and metabolic fate of individual AGE/ALE structures should be evaluated to assess whether and to what extent they are digested, absorbed and cleared by the body, and accumulate in tissues to produce any significant adverse health effect(Reference Ames451, Reference Tessier and Birlouez-Aragon527). AGE, which are absorbed as free or short peptide-bound AGE, may be cleared more rapidly and efficiently by the kidney, except when renal function is significantly impaired, than those formed endogenously, which are usually protein-bound. Studies in experimental animals fed or injected with radiolabelled protein-bound AGE and human volunteers receiving diets enriched with MRP or specific AGE have shown that absorption of ingested MRP is variable and dependent on the MRP(Reference Bergmann, Helling and Heichert464, Reference Finot and Magnenat465, Reference Tessier and Birlouez-Aragon527–Reference Somoza, Wenzel and Weiss529). Highly cross-linked AGE and melanoidins were predominantly excreted in the faeces while absorbed MRP were in general rapidly excreted by the kidney, with limited accumulation in tissues and organs(Reference Finot and Magnenat465, Reference Faist and Erbersdobler528–Reference Somoza531). Both pyrraline and pentosidine, the latter only in free form, were readily absorbed and excreted in the urine(Reference Foerster and Henle532, Reference Miyata, Ueda and Horie533). There are also reports that some MRP may be degraded by the colonic microflora(Reference Wiame, Delpierre and Collard534).
Concerning the structural requirements for AGE to interact with their receptor, there is evidence that RAGE does not bind small molecules and simple protein modifications(Reference Stern, Yan and Yan478). Penfold et al. (Reference Penfold, Coughlan and Patel535) recently showed that only serum fractions isolated from diabetic patients with nephropathy containing high-molecular-weight proteins (>50 kD) were able to stimulate RAGE but not medium- or low-molecular-weight serum fractions, in line with mechanistic studies that showed no binding of carboxymethyllysine-modified proteins to RAGE(Reference Hammes, Du and Edelstein536–Reference Thornalley, Babaei-Jadidi and Al538), and that supramolecular complexes are a prerequisite for RAGE binding(Reference Ostendorp, Leclerc and Galichet539–Reference Buetler, Latado and Leclerc542). Together this suggests that dietary AGE, even if they are absorbed from the gut, would have little effect on inflammatory processes via RAGE activation in target tissues, since it appears highly unlikely that free AGE-modified amino acids would be incorporated into nascent proteins. In addition, a few reports show that binding of AGE to RAGE is not sufficient to stimulate inflammatory signalling(Reference Thornalley, Babaei-Jadidi and Al538, Reference Buetler, Latado and Leclerc542, Reference Reznikov, Waksman and Azam543), suggesting that endotoxin contaminations of AGE preparations used in the in vitro cellular test systems may be responsible for the observed pro-inflammatory effects.
A further issue, which could complicate the interpretation of results, is the suitability of experimental design/models used in animal studies(Reference Ames451). In fact, the amount of AGE/ALE administered, and particularly the extent of their modification, might not correspond to that of AGE/ALE ingested with usual diets, due to the higher extent of modification introduced by the heating procedure used for preparing test diets in published studies(Reference Tessier and Birlouez-Aragon527). In addition, the true AGE content of many test diets has either not been determined or has been questioned by others, as in the case of the ten times concentrated caffeine-free diet cola beverage described above. In fact, others have found low to undetectable AGE levels in unconcentrated normal or diet cola(Reference Ahmed466, Reference Ahmed, Mirshekar-Syahkal and Kennish544). Similarly, the food AGE content published by Goldberg et al. (Reference Goldberg, Cai and Peppa461) and recently updated(Reference Uribarri, Woodruff and Goodman545) using a non-validated ELISA method to determine AGE content is difficult to bring in line with the Maillard reaction in general and with other data on food AGE content(Reference Ames451, Reference Tessier and Birlouez-Aragon527, Reference Assar, Moloney and Lima546).
To attribute adverse health effects to any food ingredient, it is essential that this food ingredient be tested independently, e.g. in pure form and possibly out of the food context. Unfortunately, for most published intervention studies of AGE and/or ALE, this has not been the case. The adverse health effects observed with differentially processed/heated food that result in different AGE/ALE contents have been ascribed to these protein modifications, regardless of the altered food context generated by the differential processing/heating. It may also be possible that AGE/ALE formed under these conditions might be markers of other process/heating-related alterations of food and not themselves the cause for adverse health effects. This is highlighted by the beneficial effects of melanoidins formed during heat processing of food(Reference Morales, Somoza and Fogliano547). One of the very few studies that investigated the direct effect of pure AGE by adding an AGE-modified protein v. a non-modified protein to a basal animal diet and fed to rats for 11 weeks was in fact unable to demonstrate any adverse health effects of the AGE-supplemented diet(Reference Chuyen, Arai and Nakanishi548).
A major issue is the difficulty to attribute the adverse health effects observed with thermally modified diets to their AGE/ALE content, since the possibility cannot be ruled out that other products formed such as heterocyclic amines or acrylamide(Reference Sebekova and Somoza453) or ingredients destroyed/consumed such as natural antioxidants contained in fruits and vegetables(Reference Baynes526) during cooking and food processing may be responsible for these effects. The notion that AGE may not be the agent causing postprandial inflammation is supported by a report showing that volunteers taking a single dose of either a glucose-modified, AGE-rich or a sorbitol-treated, AGE-poor casein preparation showed an identical postprandial increase in NF-κB activation in peripheral blood mononuclear cells(Reference Schiekofer, Franke and Andrassy516). There is a significant body of evidence linking food intake with postprandial inflammatory signalling. In fact, postprandial hyperglycaemia has been shown to be associated with increased NF-κB activation in patients with type 2 diabetes(Reference Rudofsky, Reismann and Schiekofer549). Moreover, an oral glucose load, or better the mean glucose excursions following an oral glucose load, were associated with urinary excretion of 8-iso PGF2α as a marker of oxidative stress within 2–3 h after the challenge(Reference Monnier, Mas and Ginet550), and oral glucose and/or high fat additively increased inflammatory markers (CRP and IL-6) and oxidative stress (nitrotyrosine), while reducing flow-mediated dilatation(Reference Ceriello, Assaloni and Da167, Reference Ceriello551) in diabetic subjects(Reference Giacco and Brownlee552). This activation during postprandial hyperglycaemia or hyperlipidaemia may be due to acute changes in markers of glycoxidative and lipoxidative stress and may partly underlie the association of postprandial derangements and cardiovascular risk(Reference Schindhelm, Alssema and Scheffer553). These observations have prompted the hypothesis that it is the sugar and lipid content of meals which is related to postprandial inflammation, oxidative stress and vascular dysfunction(Reference O'Keefe, Gheewala and O'Keefe157, Reference Ceriello, Assaloni and Da167, Reference Ceriello551, Reference Alipour, Elte and van Zaanen554). In addition, AGE precursors and AGE/ALE might simply be markers of oxidative processes and inflammation, which would be the true injurious mechanism(Reference Monnier455, Reference Sourris, Forbes and Cooper555, Reference Spiteller556). The reported beneficial effect of agents such as aminoguanidine(Reference He, Sabol and Mitsuhashi557) in preventing MRP accumulation following ingestion of high-AGE diets could be attributed to the metal-chelating and antioxidant properties of this drug(Reference Monnier455). The increased circulating and tissue AGE levels induced by AGE-enriched meals might result from oxidant stress-dependent endogenous AGE formation. The reported attenuation of inflammatory changes induced by fatty meals by treatment with antioxidants is in keeping with this view(Reference Koschinsky, He and Mitsuhashi456, Reference Negrean, Stirban and Stratmann514). Likewise, the preventive effect of benfotiamine, a transketolase activator that directs glucose substrates to the pentose phosphate pathway, might depend on the consequent blockade of endogenous dicarbonyls and AGE formation as well as of other hyperglycaemia-induced pathways(Reference Hammes, Du and Edelstein536). In addition, benfotiamine administration restores normal thiamin levels, which have been reported to be reduced in diabetic patients(Reference Thornalley, Babaei-Jadidi and Al538), and compensate for decreased thiamin ingestion with food subjected to severe thermal treatment, which is known to degrade this heat-labile vitamin(Reference Mehta, Shangari and O'Brien537, Reference Shangari, Depeint and Furrer558, Reference Shangari, Depeint and Furrer559).
Acute v. chronic exposure to AGE-enriched food could produce different effects, and, under both experimental conditions, post-meal values should be considered separately from basal fasting values. Also, the use of animals such as rodents, which are not accustomed to the intake of heated food, might result in outcomes that cannot be directly applied to humans. In addition, the effects of AGE/ALE-rich diets may be different in healthy subjects and patients with diabetes, other age-related disorders or renal insufficiency. The large variation in serum AGE levels detected in younger and older healthy subjects(Reference Uribarri, Cai and Peppa520), with some individuals showing values similar to those found in diabetic patients, would suggest that AGE might not be harmful unless a concomitant disease condition is present.
The argument that highly cooked or processed food may be responsible for adverse health effects because of their elevated AGE content may be countered by the findings that vegetarians consistently present with higher circulating AGE levels than individuals eating a Western diet(Reference Sebekova, Krajcoviova-Kudlackova and Schinzel560). Also, the recent report that obesity in children, while correlating with increased oxidative stress and inflammatory markers, showed an inverse correlation with circulating AGE levels is in disagreement with this argument(Reference Sebekova, Somoza and Jarcuskova517).
Another important issue is the reliability of methods for AGE/ALE quantification that are used by different groups(Reference Henle452, Reference Tessier and Birlouez-Aragon527, Reference Schalkwijk561). In fact, the use of antisera for quantitative immunoassays of protein-bound AGE is questionable because the specificity of the antibodies is often difficult to ascertain and monospecific antibodies are not commercially available. Furthermore, proteins used to block non-specific binding in immunoassays may also contain AGE epitopes, and thus interact with the detection system. Finally, because of steric constraints, not all AGE epitopes on the protein may be available for interaction with the antibody, and factors competing for the reaction between the anti-AGE antibody and its antigen, including anti-AGE auto-antibodies and, possibly, complement, have been demonstrated in plasma(Reference Tai and Newkirk562, Reference Turk, Ljubic and Turk563). Thus, AGE measurements with immunoassays yield only semi-quantitative results and results obtained with immunoassays in both serum samples and food should be interpreted with care. The indirect immunoassay used by Vlassara and co-workers only allows results to be expressed in units, thus making comparison with other analytical data impossible. A much better approach for the quantitative determination of specific AGE epitopes in proteins is the use of specific analytical techniques, such as liquid chromatography-MS–MS, for the analysis of specific AGE in protein hydrolysates(Reference Ahmed, Mirshekar-Syahkal and Kennish544). Application of this kind of analytical technique could lead to a more comprehensive understanding of the putative effects of AGE formed by the Maillard reaction in food as well as in the body of healthy and diabetic human subjects. In fact, conflicting carboxymethyllysine levels in different foodstuffs have been reported using either an indirect ELISA method(Reference Goldberg, Cai and Peppa461) or an ultra performance liquid chromatography-MS–MS method(Reference Assar, Moloney and Lima546).
A relevant objection to the hypothesis that dietary AGE/ALE have adverse health effects is the fact that the human organism is equipped with very effective and redundant defence mechanisms limiting the digestion and intestinal absorption of these compounds, which are also trapped in the gastrointestinal tract and detoxified in intestinal epithelia or, for those that are absorbed into the blood or lymph, in liver and other tissues. Based on the pivotal role of the gastrointestinal tract in the protection against ingested MRP, it would be important to assess whether AGE/ALE that are not absorbed have any adverse effects on the intestinal mucosa and/or colonic microflora. Preliminary studies have shown that a high-AGE diet did not affect the number or class of bifidobacteria and sulphate-reducing bacteria, which have beneficial and detrimental effects, respectively(Reference Ames451, Reference Ames, Wynne and Hofmann564). In addition, Morales et al. (Reference Morales, Somoza and Fogliano547) recently summarised the beneficial health effects of melanoidins generated during heat processing of food.
Despite all these arguments, it might be prudent to advise renal failure patients to decrease their intake of highly heated food. This may not necessarily be due to the presence of AGE/ALE in thermally treated food but rather to other factors present/absent in thermally treated food.
Finally, there may even be a ‘chemoprotective’ role of individual dietary AGE/ALE due to antioxidant(Reference Dittrich, Dragonas and Kannenkeril565, Reference Lindenmeier, Faist and Hofmann566) and anti-cancer(Reference Marko, Habermeyer and Kemeny567) properties, the former in part attributable to pronyllysine modification of lysine residues of proteins detected in certain foods including bread crust and malt(Reference Chuyen568).
Research gaps
Although there appears to be suggestive evidence for a potential role of dietary AGE in inducing low-grade chronic inflammation, insulin resistance and vascular dysfunction, the supporting proof so far has been weak, inconclusive and even contradictory. What the available studies have been able to consistently show is the relationship between dietary AGE/ALE exposure and postprandial circulating AGE levels(Reference Uribarri, Cai and Peppa520). Whether the uptake of AGE/ALE from the diet is responsible for adverse biological consequences such as chronic low-grade inflammation, vascular dysfunction or insulin resistance will require further investigations.
The following research gaps have been identified:
(1) There is a need for reliable analytical methods for food AGE with inter-laboratory cross-validations.
(2) There is a need for a food AGE database based on reliable analytical methods.
(3) There is a need for preclinical and clinical studies with pure and well-characterised AGE independent of food in both healthy individuals and diabetic patients.
(4) There is a need to discriminate the biological effects induced by protein-bound v. free well-characterised AGE.
(5) There is a need for toxicological evaluation of well-characterised AGE in animal studies.
(6) There is a need to expand investigation of the (patho)physiological role of food-derived ALE and lipid peroxidation products.
(7) There is a need to evaluate the (patho)physiological consequences of highly heated food beyond their AGE/ALE content.
(8) There is a need to balance potentially adverse health effects of AGE with the potentially beneficial health effects of melanoidins, both of which are generated by thermal treatment of food.
Macronutrients and low-grade inflammation
Fatty acids
Dietary fatty acids may affect inflammatory processes through modulation of eicosanoid metabolism, and by eicosanoid-independent mechanisms such as regulation of membrane and cytosolic signalling processes that influence the activity of transcription factors involved in inflammation(Reference Calder569, Reference Calder570). These latter transcription factors include NF-κB and PPAR-γ, both of which are sensitive to fatty acids. There is an intriguing interaction between these latter two transcription factors because PPAR-γ inhibits NF-κB activation(Reference van den Berghe, Vermeulen and Delerive571). A number of different fatty acids including saturated, monounsaturated, trans, conjugated linoleic and polyunsaturated of both n-6 and n-3 families have been investigated in the context of inflammation. They have been examined in many model systems, typically with isolated cells in culture and in animal models of inflammatory conditions, as well as in studies in human volunteers and in various patient groups. In addition, associations between intake or status of various fatty acids and inflammatory markers have been examined in human studies.
SFA
In vitro studies have suggested that SFA may promote inflammatory processes. Exposure of myotubes or adipocytes to the SFA palmitic acid (16 : 0) increased IL-6 mRNA expression and subsequent protein production, possibly via activation of NF-κB(Reference Weigert, Brodbeck and Staiger572, Reference Ajuwon and Spurlock573). Monocytes are activated directly by SFA, especially lauric acid (12 : 0), via TLR-4 and through this mechanism induce NF-κB activity(Reference Lee, Plakidas and Lee574, Reference Weatherill, Lee and Zhao575). A limited number of observational studies have investigated the relationship between SFA exposure and circulating markers of inflammation (Table 13)(Reference Lopez-Garcia, Schulze and Manson373, Reference Madsen, Skou and Hansen374, Reference Eschen, Christensen and Toft576–Reference Yli-Jama, Seljeflot and Meyer585). Fernandez-Real et al. (Reference Fernandez-Real, Broch and Vendrell577) did not see any relationship between serum SFA and CRP or IL-6 in lean individuals, while in overweight individuals, serum SFA were positively associated with IL-6 concentration, and the ratio of SFA:n-6 or n-3 PUFA was positively associated with IL-6 and CRP concentrations, respectively. A study in overweight adolescents showed positive relationships between total SFA in plasma phospholipids or cholesteryl esters and IL-6, but not CRP, concentration(Reference Klein-Platat, Drai and Oujaa579). Thus, there is general agreement between two studies in overweight subjects that SFA exposure is associated with higher IL-6 concentration. A study in lean individuals does not show this. An intervention study feeding diets rich in stearic acid (18 : 0) or in lauric, myristic (14 : 0) and palmitic acids to men aged 25–60 years for 5 weeks showed higher concentrations of CRP, fibrinogen, IL-6 and sE-selectin compared with a diet enriched in oleic acid (18 : 1n-9)(Reference Baer, Judd and Clevidence586). There are few other intervention studies chronically increasing SFA intake in human subjects and reporting inflammatory markers.
Table 13 Observational studies on the association between fatty acid intake or status and markers of low-grade inflammation

M, male; F, female; αLNA, α-linolenic acid; = , no effect on; CRP, C-reactive protein; ↓ , decreased; sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular cell adhesion molecule-1; ↑ , increased; sTNFR, soluble TNF receptor; sP-selectin, soluble P-selectin; IL-1ra, IL-1 receptor antagonist; TGF, transforming growth factor.
Trans-fatty acids
Using a subgroup of the Nurses’ Health Study, Lopez-Garcia et al. (Reference Lopez-Garcia, Schulze and Meigs580) identified significant positive associations between the intake of trans-fatty acids in the diet and the concentrations of all six inflammatory markers assessed, including CRP, IL-6 and three soluble adhesion molecules. In a 5-week intervention in healthy men, a trans-fatty acid-enriched diet resulted in higher CRP and IL-6 concentrations than diets rich in oleic acid, stearic acid or the combination of lauric, myristic and palmitic acids(Reference Baer, Judd and Clevidence586). Furthermore, the concentration of sE-selectin was higher than in all other dietary groups including the stearic acid and lauric+myristic+palmitic groups. Thus, an association study and an intervention study both demonstrate that dietary trans-fatty acids elevate the concentrations of a range of inflammatory markers including CRP, IL-6 and adhesion molecules.
Conjugated linoleic acids
In vitro and animal feeding studies have suggested marked effects of conjugated linoleic acids (CLA) on inflammation(Reference Li, Barnes and Butz587–Reference Moloney, Toomey and Noone589). However, results from human intervention studies using CLA-rich capsules are equivocal (Table 14)(Reference Mullen, Moloney and Nugent590–Reference Tricon, Burdge and Kew596). For example, two studies demonstrate that CLA, especially the trans-10, cis-12 isomer, increases CRP concentration, but not the concentrations of cytokines or soluble adhesion molecules(Reference Riserus, Basu and Jovinge593, Reference Smedman, Basu and Jovinge594). However, at least four other studies have failed to show an effect of CLA on CRP concentration (see Table 14). Duration of intervention is not likely to be an explanatory factor for differences in the findings, but, since most studies have used mixtures of CLA isomers in different proportions, the precise dose of the more potent isomer (perhaps the trans-10, cis-12 isomer) may be an explanation. The two studies showing increased CRP with CLA provided 2·1(Reference Smedman, Basu and Jovinge594) and 2·7(Reference Riserus, Basu and Jovinge593) g/d of trans-10, cis-12 CLA. The four studies showing no effect of CLA on CRP used between 0·4 and 2·5 g/d of this isomer (see Table 14). The question of whether CLA per se, or whether specific CLA isomers, increase low-grade inflammation requires further study.
Table 14 Intervention studies investigating the effect of conjugated linoleic acid intake on markers of low-grade inflammation

M, male; CLA, conjugated linoleic acid; c, cis; t, trans; ↑ , increased; CRP, C-reactive protein; = , no effect on; F, female; sICAM-1, soluble intercellular adhesion molecule-1; LT, leukotriene; sVCAM-1, soluble vascular cell adhesion molecule-1; sTNFR, soluble TNF receptor.
Linoleic and α-linolenic acids
Linoleic (18 : 2n-6) and α-linolenic (18 : 3n-3) acids constitute the majority (>95 %) of PUFA in most Western diets, with the former usually being present in an excess of approximately 10-fold over the latter. These two fatty acids are the parent n-6 and n-3 PUFA, respectively. Because of the role of linoleic acid as the precursor of arachidonic acid (20 : 4n-6), which is, in turn, the substrate for the synthesis of pro-inflammatory eicosanoids such as PGE2 and 4-series leukotrienes, it is widely considered that elevated n-6 and low n-3 fatty acids (i.e. a high n-6:n-3 PUFA ratio) in the diet will promote inflammation. However, available evidence does not support this contention.
Dietary intakes of linoleic acid were not associated with CRP, IL-6, sTNFR1 or sTNFR2 concentrations in subgroups of the Physicians’ Health Study and the Nurses’ Health Study(Reference Pischon, Hankinson and Hotamisligil583). The concentration of linoleic acid in blood lipids(Reference Ferrucci, Cherubini and Bandinelli578, Reference Klein-Platat, Drai and Oujaa579) or granulocytes(Reference Madsen, Skou and Hansen374) was not associated with CRP or IL-6 concentration, although a large Swedish study reported an inverse association between linoleic acid in cholesteryl esters and CRP concentration(Reference Petersson, Basu and Cederholm582). Linoleic acid makes a major contribution to the fatty acids within blood lipids, especially cholesteryl esters, where it is the predominant n-6 PUFA. Total n-6 PUFA in serum fatty acids in overweight, but not in lean, subjects were inversely associated with IL-6 but not CRP concentration(Reference Fernandez-Real, Broch and Vendrell577). The ratio of SFA:n-6 PUFA in serum lipids or in plasma phospholipids was positively associated with IL-6 but not CRP concentration in overweight subjects(Reference Fernandez-Real, Broch and Vendrell577, Reference Klein-Platat, Drai and Oujaa579). This suggests that decreasing SFA status while increasing n-6 PUFA status might reduce low-grade inflammation. An intervention study in thirty-eight healthy male and female volunteers (mean age 27 years) compared diets with 18 % energy from oleic acid and 4 % from linoleic acid or 4 % energy from oleic acid and 12 % from linoleic acid; plasma sICAM-1 concentrations were not different between these two groups(Reference Turpeinen, Basu and Mutanen597), suggesting that exchange of oleic acid for linoleic acid, while keeping SFA intake constant, does not affect this marker of inflammation.
Dietary α-linolenic acid intakes were not associated with CRP, IL-6, sTNFR1 or sTNFR2 concentrations in one study on subgroups of the Physicians’ Health Study and the Nurses’ Health Study(Reference Pischon, Hankinson and Hotamisligil583). In a second study on another subgroup of the Nurses’ Health Study, α-linolenic acid intakes were not associated with CRP, sICAM-1, sE-selectin or sTNFR2 concentrations but were associated with lower IL-6 and sVCAM-1 concentrations(Reference Lopez-Garcia, Schulze and Manson373). The concentration of α-linolenic acid in blood lipids(Reference Ferrucci, Cherubini and Bandinelli578, Reference Klein-Platat, Drai and Oujaa579, Reference Petersson, Basu and Cederholm582) or granulocytes(Reference Madsen, Skou and Hansen374) was not associated with CRP or IL-6 concentration. Data from an Italian study showed no association between α-linolenic acid in plasma fatty acids and several cytokines including IL-6 and TNF-α, but there was an inverse association with CRP(Reference Ferrucci, Cherubini and Bandinelli578). These association studies suggest a modest anti-inflammatory effect of α-linolenic acid.
Several intervention studies have involved high α-linolenic acid intakes usually by providing flaxseed oil in capsules or in liquid form or foodstuffs made using flaxseed oil(Reference Burdge and Calder598). Frequently, these studies have used a control group with a high intake of linoleic acid, with the comparison essentially being replacement of linoleic acid with α-linolenic acid. Thus, the focus of these studies is essentially to explore the importance of the n-6:n-3 PUFA ratio of the diet. If linoleic and α-linolenic acids have similar effects on low-grade inflammation, then studies exchanging one of these fatty acids with the other would be likely to see little effect. Table 15 summarises intervention studies with α-linolenic acid that have measured markers of low-grade inflammation as an outcome. Findings are inconsistent with a number of studies identifying the effects of α-linolenic acid on some markers and not others, and some studies finding no effects (Table 15)(Reference Bemelmans, Lefrandt and Feskens599–Reference Zhao, Etherton and Martin606). Many studies have used very high intakes of α-linolenic acid, relative to typical habitual intakes. The study of Paschos et al. (Reference Paschos, Rallidis and Liakos601) is enlightening because it showed that α-linolenic acid (8·1 g/d for 12 weeks) was less effective (i.e. induced fewer and smaller effects) against a more healthy, than against a less healthy, background diet. Thus, dose, duration, sample size and the nature of the background diet are possible contributors to the varied findings of studies with α-linolenic acid. However, what is apparent from these observations is that a substantial increase in the intake of α-linolenic acid can decrease low-grade inflammation as indicated by circulating CRP, IL-6 or soluble adhesion molecules (see Table 15). The study of Zhao et al. (Reference Zhao, Etherton and Martin606) provides further insight: the change in CRP and sVCAM-1 concentrations was correlated with the change in the concentrations of different serum n-3 PUFA (α-linolenic acid, EPA, docosapentaenoic acid (22 : 5n-6) and DHA) that had occurred. The only significant relationships were inverse and between the change in EPA status and the changes in CRP and sVCAM-1 concentrations. This suggests that the anti-inflammatory effect seen with the very high intake of α-linolenic acid in this study is due to the conversion of α-linolenic acid to EPA. A suitable conclusion may be that high intakes of α-linolenic are anti-inflammatory acting via the α-linolenic acid derivative, EPA.
Table 15 Intervention studies investigating the effect of α-linolenic acid (αLNA) intake on markers of low-grade inflammation

M, male; F, female; = , no effect on; CRP, C-reactive protein; sL-selectin, soluble L-selectin; sP-selectin, soluble P-selectin; PAI, plasminogen activator inhibitor; sICAM-1, soluble intercellular adhesion molecule-1; ↓ , decreased; sVCAM-1, soluble vascular cell adhesion molecule-1; SAA, serum amyloid A; MCSF, macrophage colony-stimulating factor.
Several of the intervention studies with α-linolenic acid have involved a group consuming a high intake of linoleic acid, frequently as the control for the high α-linolenic acid intake. These groups provide some information about the impact of linoleic acid on low-grade inflammation. The study of Rallidis et al. (Reference Rallidis, Paschos and Liakos603, Reference Rallidis, Paschos and Papaioannou604) involved a control group consuming about 11 g/d of linoleic acid in addition to the dietary intake; this approximately doubled linoleic acid intake. The increase in linoleic acid intake did not alter the concentrations of CRP, IL-6, SAA, sICAM-1 or sE-selectin, but the concentration of sVCAM-1 was decreased(Reference Rallidis, Paschos and Liakos603, Reference Rallidis, Paschos and Papaioannou604). In the study of Paschos et al. (Reference Paschos, Zampelas and Panagiotakos602), the control group consumed 11·2 g/d of linoleic acid on top of their normal intake, which was approximately 11 g/d. This doubling of linoleic acid intake did not affect TNF-α or adiponectin concentrations. These studies did not alter any aspect of diet but required subjects to consume oil providing linoleic or α-linolenic acids on top of the normal diet. Thus, these studies show that markedly increasing linoleic acid intake in those consuming on average about 10 g/d does not increase low-grade inflammation.
The study of Zhao et al. (Reference Zhao, Etherton and Martin606) included a high linoleic acid intervention group (13 % energy) and the control group consumed what is described as an ‘average American diet’. This provided 12·7 % of energy from SFA, 7·7 % from linoleic acid and 0·8 % from α-linolenic acid. The corresponding values for the linoleic and α-linolenic acid rich diets were 8·5, 12·6 and 3·6 and 8·2, 10·5 and 6·5, respectively. Thus, the linoleic acid-rich diet contained more α-linolenic acid than the control diet, while the α-linolenic acid diet contained more linoleic acid than the control diet and almost as much linoleic acid as the linoleic acid diet. Essentially, what this means is that the linoleic acid diet is examining the effect of replacing some SFA with the combination of linoleic and α-linolenic acids and that the α-linolenic acid diet is examining the effect of replacing some linoleic acid with α-linolenic acid. In the linoleic acid group, the concentrations of CRP and sICAM-1 decreased by 45 and 25 %, respectively; sVCAM-1 and sE-selectin also fell (by 15 and 7 %) but these changes were not significant. Thus, replacing about one-third of SFA with linoleic acid plus α-linolenic acid decreases low-grade inflammation. This is consistent with the associations described above and confirms that relative to SFA, the combination of linoleic and α-linolenic acids is anti-inflammatory. The changes in inflammatory markers seen with the α-linolenic acid diet were greater than with the linoleic acid diet (75, 80, 37·5 and 12 % decreases in CRP, sVCAM-1, sICAM-1 and sE-selectin concentrations, respectively). Since this diet is effectively replacing some linoleic acid with α-linolenic acid, relative to the amounts in the linoleic acid diet, these results suggest that α-linolenic acid is more potent than linoleic acid with regard to reducing inflammation.
The studies just described have focused on increasing intake of linoleic and α-linolenic acids to study their effects. The study of Liou et al. (Reference Liou, King and Zibrik607) used a different approach. They kept the amount of α-linolenic acid in the diet of men aged 20–45 years constant over the study period at about 1 % of energy. Linoleic acid intake was either 10·5 or 3·8 % of energy. Saturated fat was also constant across the two diets at about 8 % of energy. The manipulation of linoleic acid was at the expense of oleic acid (17 or 10 % of energy). The intervention duration was 4 weeks and a random order, cross-over design was used. CRP and IL-6 concentrations were not different after 4 weeks on either diet. This finding is in accordance with that of Turpeinen et al. (Reference Turpeinen, Basu and Mutanen597).
Arachidonic acid
Arachidonic acid intake in the diet is low relative to that of its metabolic precursor linoleic acid (approximately 500 mg/d v. approximately 11 g/d, respectively). Nevertheless, arachidonic acid is the most prevalent n-6 PUFA and PUFA in the membranes of inflammatory cells and other cells that might be involved in low-grade inflammation such as endothelial cells and platelets. This reflects the important functional role of arachidonic acid as a precursor of the eicosanoid family of lipid mediators; this family includes the 2-series PG and the 4-series leukotrienes. Since these eicosanoids are classically associated with inflammatory processes and are targeted by common anti-inflammatory therapies, it is generally considered that arachidonic acid will enhance inflammation. However, observations that classical pro-inflammatory mediators such as PGE2 can also exert anti-inflammatory effects and that arachidonic acid gives rise to anti-inflammatory mediators such as lipoxin A4 have started to challenge the earlier view(Reference Calder608). Several studies have examined the association between arachidonic acid status and markers of low-grade inflammation. There was no association between arachidonic acid in granulocytes and CRP concentration(Reference Madsen, Skou and Hansen374). Serum free arachidonic acid was not associated with sICAM-1 or sE-selectin concentrations and was actually inversely associated with sVCAM-1 concentration(Reference Yli-Jama, Seljeflot and Meyer585). Ferrucci et al. (Reference Ferrucci, Cherubini and Bandinelli578) reported no association between arachidonic acid in plasma and CRP, TNF-α, IL-1β, IL-10 and sIL-6R concentrations, while there was an inverse association with IL-6 and IL-1ra concentrations and a positive association with TGF-β concentration. These observations suggest either that plasma arachidonic acid has little impact on low-grade inflammation (does not affect CRP or TNF-α) or that it is anti-inflammatory (lowers IL-6; increases TGF-β).
There are very few intervention studies with arachidonic acid reporting on low-grade inflammation. In an uncontrolled study, Kelley et al. (Reference Kelley, Taylor and Nelson609) reported higher granulocyte numbers in the blood of a group of ten healthy men (aged 20–38 years) taking a supplement of 1·5 g/d of arachidonic acid for 100 d compared with numbers after a run-in diet providing 200 mg/d of arachidonic acid. In another small, but controlled, study, eight subjects aged 55–75 years consumed capsules providing 700 mg/d of arachidonic acid for 12 weeks(Reference Thies, Miles and Nebe-von-Caron605); there was no effect on plasma sVCAM-1, sICAM-1 or sE-selectin concentrations.
Marine-derived long-chain n-3 PUFA
The long-chain n-3 PUFA EPA and DHA are found in seafood, especially oily fish. They are also present in fish oils and in certain algal oils; in some preparations, the fatty acids are in a more concentrated form than in natural fish oils. In fish oils, the fatty acids are in the TAG form, but other forms of long-chain n-3 PUFA are also available, for example, as phospholipids or ethyl esters. Increased intake of long-chain n-3 PUFA results in increased proportions of those fatty acids in inflammatory cell phospholipids(Reference Yaqoob, Pala and Cortina-Borja610–Reference Calder614). The incorporation of EPA and DHA into human inflammatory cells is partly at the expense of arachidonic acid, resulting in less substrate available for the synthesis of the classic inflammatory eicosanoids such as PGE2. Through altered eicosanoid production, n-3 PUFA could affect inflammation and inflammatory processes, although they also exert non-eicosanoid-mediated actions on cell signalling and gene expression. The effects of long-chain n-3 PUFA have been examined in many model systems and findings from cell-culture systems and from animal models are generally consistent in identifying anti-inflammatory actions(Reference Calder569). Furthermore, clinical trials have demonstrated anti-inflammatory effects and clinical benefit from fish oil administration in diseases with a frank inflammatory basis including rheumatoid arthritis(Reference Calder615), inflammatory bowel diseases(Reference Calder616) and childhood asthma(Reference Kremmyda, Vlachava and Noakes617).
Data from subgroups of the Physicians’ Health Study and the Nurses’ Health Study showed inverse associations between the dietary intake of EPA+DHA and concentrations of CRP, sTNFR1 and sTNFR2(Reference Pischon, Hankinson and Hotamisligil583), and CRP, sICAM-1, sVCAM-1 and sE-selectin(Reference Lopez-Garcia, Schulze and Manson373). The concentration of either EPA or DHA in granulocyte membranes was inversely associated with CRP concentration in one study(Reference Madsen, Skou and Hansen374); the effect of DHA was stronger than that of EPA. Serum non-esterified EPA and DHA were both inversely associated with concentrations of sVCAM-1 and sICAM-1 in patients at risk of CHD(Reference Yli-Jama, Seljeflot and Meyer585); EPA was also inversely associated with sE-selectin concentration. Plasma cholesteryl ester EPA was inversely associated with CRP concentration in overweight subjects(Reference Klein-Platat, Drai and Oujaa579). In an elderly Italian population, plasma EPA was inversely associated with IL-6 concentration and positively associated with the concentrations of the anti-inflammatory cytokines IL-10 and TGF-β(Reference Ferrucci, Cherubini and Bandinelli578). Furthermore, plasma DHA was inversely associated with IL-6 and TNF-α concentrations and was also positively associated with the concentrations of IL-10 and TGF-β(Reference Ferrucci, Cherubini and Bandinelli578). Thus, observational studies suggest that both EPA and DHA are anti-inflammatory.
The ready availability of fish oil capsules has facilitated numerous supplementation studies of long-chain n-3 PUFA in various subject groups; these are summarised in Table 16(Reference Jellema, Plat and Mensink164, Reference Thies, Miles and Nebe-von-Caron605, Reference Abe, El-Masri and Kimball618–Reference Yusof, Miles and Calder646). Studies have shown that long-chain n-3 PUFA lower the concentrations of CRP(Reference Browning, Krebs and Moore621, Reference Ciubotaru, Lee and Wander624, Reference Rasic-Milutinovic, Perunicic and Pljesa639), IL-6(Reference Browning, Krebs and Moore621, Reference Ciubotaru, Lee and Wander624, Reference Rasic-Milutinovic, Perunicic and Pljesa639), TNF-α(Reference Rasic-Milutinovic, Perunicic and Pljesa639), IL-18(Reference Troseid, Arnesen and Hjerkinn643), sICAM-1(Reference Eschen, Christensen and De625, Reference Hjerkinn, Seljeflot and Ellingsen628, Reference Yamada, Yoshida and Nakano645, Reference Yusof, Miles and Calder646), sVCAM-1(Reference Thies, Miles and Nebe-von-Caron605, Reference Yamada, Yoshida and Nakano645) and sE-selectin(Reference Abe, El-Masri and Kimball618) in various subject/patient groups (see Table 16). For example, one study showed an increase in adiponectin concentration when a weight-loss programme and 4·2 g/d EPA+DHA were combined in overweight, insulin-resistant women(Reference Krebs, Browning and McLean632). Thus, there is quite a lot of evidence for anti-inflammatory effects of supplemental long-chain n-3 PUFA. In a group of overweight women with type 2 diabetes, 8 weeks of a moderate dose of long-chain n-3 PUFA (1·8 g/d of EPA+DHA) decreased adiposity and reduced expression of a number of inflammation-related genes in the subcutaneous adipose tissue(Reference Kabir, Skurnik and Naour630). The parallelism between the down-regulation of these genes and the reduction in adiposity and adipocyte diameter by n-3 PUFA treatment suggests a positive relationship between adipose cell size and adipose tissue inflammation, agreeing with other observations(Reference Cancello, Henegar and Viguerie31, Reference Weisberg, McCann and Desai66). Also these findings in type 2 diabetic women are paralleled by those from rodent studies: 6 weeks of n-3 PUFA supplementation prevented adipose tissue inflammation induced by a high-fat diet(Reference Todoric, Loffler and Huber647), and the presence of n-3 PUFA in the diet of long-term insulin-resistant, sucrose-fed rats decreased adipocyte diameter(Reference Rossi, Lombardo and Lacorte648) and significantly reduced several inflammation-related genes (unpublished results, Guerre-Millo M, Naour N, Lombardo Y, Clement K, Rizkalla S). These studies suggest that reducing adiposity with n-3 PUFA could decrease adipose tissue inflammation and macrophage infiltration. The beneficial effects of n-3 PUFA may be linked to local blunting of adipose tissue inflammation.
Table 16 Intervention studies investigating the effect of marine n-3 PUFA intake on markers of low-grade inflammation

M, male; FO, fish oil; = , no effect on; IL-1ra, IL-1 receptor antagonist; F, female; EE, ethyl ester; sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular cell adhesion molecule-1; ↓ , decreased; sE-selectin, soluble E-selectin; ↑ , increased; sP-selectin, soluble P-selectin; PAI-1, plasminogen activator inhibitor-1; CRP, C-reactive protein; sTNFR, soluble TNF receptor; SAA, serum amyloid A.
Despite the large number of positive studies with long-chain n-3 PUFA, there are a number of studies that have failed to replicate these findings (see Table 16). Furthermore, two early studies showed an enhancement of selected inflammatory markers following long-chain n-3 PUFA administration(Reference Johansen, Seljeflot and Hostmark629, Reference Seljeflot, Arnesen and Brude642). Seljeflot et al. provided 4·8 g/d of EPA+DHA in ethyl ester form to hyperlipidaemic male smokers for 6 weeks, while Johansen et al. provided 5·1 g/d of EPA+DHA in ethyl ester form to patients with CHD for 24 weeks. Both studies identified an increase in sVCAM-1 ( < 10 %) and sE-selectin (20 %) concentrations, with no effect on sP-selectin and a decrease in von Willibrand factor and thrombomodulin concentrations. In both cases, the authors ascribed the effect of EPA+DHA on sVCAM-1 and sE-selectin to increased oxidant stress in these subjects.
Thus, although the overwhelming view is that EPA+DHA given at sufficient doses are anti-inflammatory, the evidence from measurements of markers of low-grade inflammation is not entirely consistent. The lack of consistency may be related to differences in: duration of treatment; sample size; characteristics of the populations studied (e.g. age, healthy v. diseased, type of disease, smokers v. non-smokers); background diet; dose of EPA+DHA used; relative contribution of EPA and DHA, since they may have different anti-inflammatory potencies; chemical formulation (e.g. TAG v. ethyl ester); degree of oxidative stress present. One other factor that has been recently identified is genetic differences among individuals, which may have an impact on the ability of n-3 PUFA to exert an anti-inflammatory effect. This was first identified by Grimble et al. (Reference Grimble, Howell and O'Reilly649) who showed that the ability of fish oil to lower the LPS-stimulated production of TNF-α by blood mononuclear cells was determined in part by polymorphisms within the TNF-α and TNF-β genes. Another example of such an interaction was identified by Shen et al. (Reference Shen, Arnett and Peacock650). They first identified that the IL-1β 6054 G>A SNP was significantly associated with CRP and adiponectin concentrations and with the prevalence of the metabolic syndrome among a group of 1120 men and women with a mean age of 49 years. There was also a significant interaction between this polymorphism and erythrocyte membrane n-3 PUFA content. Among subjects with low erythrocyte n-3 PUFA content (below the median), the 6054 G allele was associated with increased risk of the metabolic syndrome (OR 3·29, 95 % CI 1·49, 7·26 for GG and OR 1·95, 95 % CI 0·85, 4·46 for GA) compared with the AA genotype, but there were no significant genotype associations among subjects with high erythrocyte n-3 PUFA content (above the median). The results suggest that IL-1β genetic variants are associated with measures of chronic low-grade inflammation and the risk of the metabolic syndrome, and that genetic influences were more evident among subjects with low erythrocyte n-3 PUFA status and so, most probably low n-3 PUFA intake.
Carbohydrates
Acute postprandial effects of hyperglycaemia
Hyperglycaemia, elevated TAG/NEFA and hyperinsulinaemia are events that are mutually inherent to the initial development of a diabetic state. The effects of the individual events are difficult to study since they always present together. However, some observations have suggested that hyperglycaemia and hypertriacylglycerolaemia/elevated NEFA levels induce independent effects and, when present together, act synergistically to generate oxidative stress, inflammation, impaired endothelial function and vascular disease(Reference Ceriello, Quagliaro and Piconi651–Reference Kempf, Rose and Herder653). Oxidative stress is known to induce damage to cell membranes, internal cell structures and DNA as well as to induce inflammatory responses (Fig. 8) (Reference Giugliano, Ceriello and Esposito654). Ceriello et al. (Reference Ceriello, Taboga and Tonutti652) observed that after meals LDL oxidation increases and that this phenomenon is correlated with the degree of hyperglycaemia. This clearly points to hyperglycaemia-induced free radical production that has an impact on a broad range of metabolic events. NO, a potent vasodilator, is assumed to play a key role in this respect. It has been suggested that hyperglycaemia leads to an increased oxidation of NO, thereby reducing NO levels which leads to impairment in vasodilatation(Reference Davidson and Duchen655). Indeed, alloxan diabetic rats are observed to have reduced NO levels in blood(Reference Mohan and Das656), and in human subjects, acute hyperglycaemia attenuated endothelium-dependent vasodilation(Reference Williams, Goldfine and Timimi657). Postprandial hyperglycaemia is also correlated with impaired myocardial perfusion in diabetic patients(Reference Scognamiglio, Negut and De Kreutzenberg658). The fact that such a decrease can be prevented by supplying various antioxidants such as vitamin C, vitamin E and α-lipoic acid(Reference Jacob, Ruus and Hermann659–Reference Evans, Goldfine and Maddux662) or l-arginine(Reference Mohan and Das656, Reference Giugliano, Marfella and Coppola663), the precursor of NO, suggests that increased NO oxidation and the related NO drain have an impact on cells of the vasculature. This is supported by other evidence. Restoration of NO availability results in normalisation of endothelial function as well as insulin sensitivity(Reference Jacob, Ruus and Hermann659, Reference Caballero660). During an oral glucose tolerance test, plasma antioxidant status, measured as total plasma radical trapping capacity, significantly decreased in normal as well as in diabetic individuals. The consumption of a glycaemic meal increases oxidative stress and reduces antioxidant defences, with the increase being significantly greater with higher levels of hyperglycaemia(Reference Ceriello, Bortolotti and Motz169, Reference Ceriello, Bortolotti and Motz664, Reference Ursini, Zamburlini and Cazzolato665). Acute post-meal hyperglycaemia was observed to induce the formation of nitrotyrosine, a marker of oxidative stress, in healthy, non-overweight individuals(Reference Marfella, Quagliaro and Nappo666). It has been hypothesised that acute hyperglycaemia may induce a drain of vitamin C from cells because vitamin C and glucose share a common transport system(Reference Price, Price and Reynolds667) and oxidative stress leads to a use of intracellular vitamin C. Accordingly, Chen et al. (Reference Chen, Hutchinson and Pecoraro668) provided evidence in vitro that acute hyperglycaemia leads to a significant decrease in leucocyte vitamin C content. Evans et al. (Reference Evans, Goldfine and Maddux662) developed a unique oxidative stress hypothesis suggesting that chronic elevation of hyperglycaemia (and NEFA) induces an activation of the NF-κB, p38 MAPK and NH2-terminal Jun kinase/stress-activated protein kinase pathways. This happens along with the activation of the RAGE, protein kinase C and sorbitol stress pathways. The authors suggest that these events play a key role in causing late complications in type 1 and type 2 diabetes, going along with insulin resistance and impaired insulin secretion in type 2 diabetes. Evans et al. (Reference Evans, Goldfine and Maddux662) provided evidence that elevated glucose causes oxidative stress due to increased production of mitochondrial ROS, non-enzymatic glycation of proteins and glucose auto-oxidation. They also pointed out that elevated NEFA can cause oxidative stress due to increased mitochondrial uncoupling and β-oxidation, the latter leading to an increased production of ROS and to an activation of stress-sensitive signalling pathways, which in turn impair insulin secretion and action. Thus, oxidative stress induced by acute and chronic elevations in glucose and NEFA plays a key role in causing insulin resistance and β-cell dysfunction. A range of studies has shown that acute and lasting hyperglycaemia leads to an elevation in dicarbonyls and protein glycation (measured as fructosamine or HbA1c). Glycosylated proteins, as well as their metabolites, AGE, which bind to the AGE receptor RAGE, induce a cascade of signals that initiate an inflammatory response(Reference Beisswenger, Howell and O'Dell669). Accordingly, pro-inflammatory transcription factors, inflammatory markers including CRP, IL-6, IL-8, TNF-α and matrix metalloproteinase and markers of endothelial dysfunction VCAM-1, ICAM-1 and E-selectin have been observed to be elevated in response to acute and persistent hyperglycaemia(Reference Bierhaus, Humpert and Morcos474, Reference Bierhaus, Rudofsky and Humpert475, Reference Negrean, Stirban and Stratmann514, Reference Vlassara, Cai and Crandall521, Reference Kempf, Rose and Herder653, Reference Evans, Goldfine and Maddux662, Reference Cai, He and Zhu670, Reference Kempf, Rose and Herder671).

Fig. 8 Schematic representation of the general mechanisms by which hyperglycaemia can affect inflammation. PKC, protein kinase C. Reproduced with permission from Giugliano et al. (Reference Giugliano, Ceriello and Esposito654).
Glycaemic index, glycaemic load and inflammation
Dietary glycaemic index (GI) and glycaemic load (GL) were both positively associated with plasma CRP concentration in 18 137 healthy women aged >45 years without diagnosed diabetes(Reference Levitan, Cook and Stampfer672). In another study in 974 subjects aged 42–87 years, dietary GI was positively associated with CRP concentration(Reference Du, van der and van Bakel673). Qi et al. (Reference Qi, Rimm and Liu315) examined the associations of dietary GI and GL with plasma adiponectin among 780 diabetic men from the Health Professionals’ Follow-Up Study. After adjustment for other factors, dietary GI and GL were both significantly inversely associated with plasma adiponectin concentration in a dose-dependent fashion. Adiponectin levels were 13 % lower in the highest quintile of dietary GI than in the lowest quintile. For dietary GL, adiponectin levels were 18 % lower in the highest quintile than in the lowest. Several intervention studies have examined the impact of dietary GI or GL on markers of chronic low-grade inflammation. In one study, thirty-four healthy overweight adults aged 24–42 years received energy-restricted diets with high or low GL for 6 months(Reference Pittas, Roberts and Das674); more subjects in the low GL group showed a decline in serum CRP concentration than in the high GL group. In another study, Shikany et al. (Reference Shikany, Phadke and Redden675) found no differences in CRP, TNF-α, IL-6, fibrinogen, sTNFR2 or PAI-1 concentrations when overweight or obese men consumed diets with high or low GI for 4 weeks. Likewise, 11-week interventions of high- or low-GL diets in fifteen overweight subjects had no differential effects on CRP, TNF-α, IL-6 or MCP-1 concentrations(Reference Vrolix and Mensink676). Thus, evidence from large observational studies is highly suggestive that there is a positive association between GI/GL and low-grade inflammation, but intervention trials do not support this convincingly, perhaps because of the small size of most of the latter trials.
Dietary fibre
In NHANES 1999–2000 in the USA, dietary fibre intake was inversely associated with serum CRP concentrations (OR 0·49 for 32 g/d v. 5·1 g/d) in diabetic women (n 3920)(Reference Ajani, Ford and Mokdad677), as well as in subjects with diabetes, hypertension and obesity (n 7891)(Reference King, Mainous and Egan678). Similar findings were reported in a cross-sectional study with healthy subjects (n 205) and in subjects with metabolic disorders (n 1653) in Italy(Reference Bo, Durazzo and Guidi679). In the Seasonal Variation of Blood Cholesterol Levels Study involving 524 subjects, who had multiple measurements of CRP and dietary intake, dietary total fibre and soluble and insoluble fibre were all inversely related to CRP concentration in the regression models adjusted for several traditional cardiovascular risk factors, current infection status and season of year(Reference Ma, Griffith and Chasan-Taber680). In contrast, the same authors observed no correlation between dietary fibre intake and blood CRP concentrations with a single measurement at baseline in the Women's Health Initiative Observational Study(Reference Ma, Hebert and Li681). However, there was an inverse association between fibre intake and IL-6 and sTNFR2 concentrations in this female population. In a study with diabetic males (n 780, Health Professionals’ Follow-Up Study), cereal fibre was positively associated with adiponectin levels after controlling for a number of confounding factors(Reference Qi, Rimm and Liu315). King et al. (Reference King, Egan and Woolson682) conducted an intervention study with thirty-five lean normotensive and seventeen obese hypertensive adults that involved a randomised cross-over design. The two intervention diets constituted either the Dietary Approaches to Stop Hypertension (DASH) diet (naturally high in fibre i.e. 30 g fibre/d) or a fibre-supplemented usual diet (30 g psyllium fibre/d) each for a 3-week period. Both diets caused a reduction in CRP concentration (14 and 18 %, respectively), although this was significant only in lean normotensive subjects in either intervention arm. The data generally support that dietary fibre intake is associated with reduced low-grade inflammation.
Milk peptides
Increased consumption of dairy products has been shown to have beneficial effects on plasma CRP and adiponectin concentrations in obese subjects(Reference Zemel and Sun683). This effect has been suggested to be partly explained by the intake of dairy protein-derived peptides. Indeed, the casein-derived peptides Ile-Pro-Pro and Val-Pro-Pro slightly, though not significantly, lowered CRP concentration in hypertensive subjects after 10 weeks(Reference Jauhiainen, Vapaatalo and Poussa684). Whey protein-derived peptides lowered CRP concentration (after 6 weeks) in one study in hypertensive subjects(Reference Pins and Keenan685), but not in another(Reference Lee, Skurk and Hennig686). Minor dairy proteins and peptides, especially lactoferrin, also show anti-inflammatory effects in different models(Reference Crouch, Slater and Fletcher687–Reference Legrand, Elass and Carpentier689), and hence there is potential for a variety of milk peptides to have anti-inflammatory effects, but there are no or insufficient human studies to allow evaluation of their efficacy.
Micronutrients and phytochemicals
Iron
The topic of Fe in relation to low-grade chronic inflammation is complex. Part of the complexity is related to the fact that Fe status assessment is confounded in the presence of inflammation(Reference Ahluwalia690–Reference Baynes692). Fe status assessment relies on a battery of laboratory tests spanning various stages of Fe deficiency. These tests include: serum ferritin, which generally indicates body Fe stores; soluble receptor of transferrin in serum (transferrin receptor) reflecting tissue Fe; serum transferrin, total Fe-binding capacity and transferrin saturation, which indicate Fe-deficient erythropoiesis; finally, red cell indices namely mean cell volume, Hb and haematocrit, which are considered functional Fe indices. Others have suggested the ratio of transferrin receptor:serum ferritin as an index of total body Fe stores(Reference Cook, Flowers and Skikne693). Most tests of Fe status, however, are affected in response to subclinical inflammation and infections(Reference Ahluwalia690–Reference Baynes692). Thus, it becomes difficult to study the relationship of Fe with any outcome of interest when inflammation is present. Recent studies have suggested that serum transferrin receptor remains unaffected in the presence of inflammation or infectious disease (reviewed in Ahluwalia(Reference Ahluwalia690) and Northrop-Clewes(Reference Northrop-Clewes694)), thus allowing the examination of the role of Fe in conditions with an underlying inflammatory component.
Deficiency v. excess
Fe has been called a double-edged sword, as both Fe deficiency and excess can have deleterious effects(Reference Weiss695). Fe is important in immune/inflammatory responses including neutrophil activation, macrophage effector functions, and T-helper cell type-1 and -2 (Th1/Th2) patterns (reviewed in Weiss(Reference Weiss695)). Fe deficiency is associated with alterations in the immune response and increased risk of infections(Reference Weiss695–Reference de Silva, Atukorala and Weerasinghe697). On the other hand, because Fe is a redox-active transition metal, it may contribute to the production of ROS, oxidative stress and inflammation(Reference Wood698, Reference Wagener, Volk and Willis699), and thus Fe excess can augment the risk for diabetes and CVD. It is important to indicate that ROS are produced by free, and not bound, Fe(Reference McCord700) and the body has evolved a metabolic system that minimises the availability of free Fe(Reference Wood698). Most Fe in the body is not free, and is bound to proteins such as ferritin, transferrin and Hb; thus, serum ferritin, transferrin, transferrin saturation and Hb measurements do not reflect the availability of ‘free’ or ‘labile’ Fe that is implicated in the production of ROS. In recent years, the measurement of non-transferring-bound labile Fe(Reference Lee and Jacobs701) has been introduced; however, few studies have utilised this measurement to date due to technical, cost and standardisation issues.
Iron status and low-grade inflammation
There is some epidemiological evidence that higher Fe intake, particularly that of haem Fe, and higher Fe status are associated with increased risk of type 2 diabetes, atherosclerosis and CHD(Reference Rajpathak, Ma and Manson702–Reference Ahluwalia, Genoux and Ferrieres714), although not all of the literature is consistent. It is not clear whether the association of Fe intake and status with diabetes or CVD is mediated through the effects of Fe on inflammatory pathways. In a small study with thirty-one carbohydrate-intolerant patients, quantitative phlebotomy was used to induce Fe depletion to near-deficiency levels(Reference Facchini and Saylor715). The induced Fe deficiency was associated with reductions in several CVD risk factors as well as in the inflammatory marker fibrinogen. While uncertainty exists as to whether Fe plays a causative role in the aetiology of low-grade inflammation and its associated pathologies, past and emerging evidence indicates that chronic low-grade inflammation is associated with poor Fe status in obese persons(Reference Selzer and Mayer716–Reference Yanoff, Menzie and Denkinger721). A concomitant improvement in Fe status (as measured by transferrin saturation) and decrease in inflammation (CRP and orosomucoid concentrations) has been observed after bariatric surgery intervention in morbidly obese women(Reference Anty, Dahman and Iannelli722). In total, emerging evidence indicates that the low-grade inflammation of obesity may be associated with low Fe status; however, further investigation of this relationship is warranted.
A small number of studies have investigated the effects of increasing total Fe or haem Fe intake on markers of inflammation(Reference Hodgson, Ward and Burke723–Reference Schumann, Kroll and Weiss725). In a small study involving three healthy volunteers who received 120 mg Fe/d for a week, there were no changes in circulating CRP concentration or leucocyte counts, or in urinary neopterin concentration(Reference Schumann, Kroll and Weiss725). Furthermore, postpartum Fe supplementation (80 mg/d) for 12 weeks in non-anaemic Fe-deficient women did not significantly alter CRP concentration or leucocyte counts(Reference Krafft, Perewusnyk and Hanseler726). In a small study in 8–11-year-old Guatemalan children who received twice the recommended daily amounts of Fe for 8 weeks (n 20) or placebo (n 20), no differences in CRP or orosomucoid concentrations were noted, although α-1 antichymotrypsin levels were increased with the Fe supplement(Reference Rosales, Kang and Pfeiffer724). In another study examining the effect of increasing lean red meat intake, participants were either assigned to a control group (maintain their usual diet) or to partially replace energy from carbohydrate-rich foods with 200 g/d of lean red meat for 8 weeks(Reference Hodgson, Ward and Burke723). In this short intervention, increased red meat intake was not associated with increased oxidative stress or inflammation. Taken together, the evidence from these limited studies examining the effect of Fe supplementation or increased red meat intake on inflammatory markers suggests no major effect on inflammation; however, there is a need for further larger well-designed studies to clarify this effect.
Vitamin D
Various immune cells including monocytes/macrophages, dendritic cells, T-cells and B-cells can convert inactive vitamin D3 to its active form (1,25(OH)2D3), and these cells can also respond to the active vitamin D via its receptor which they express(Reference Froicu, Zhu and Cantorna727). It seems likely that vitamin D plays a paracrine modulatory role in the immune/inflammatory system(Reference Baeke, van and Gysemans728). Although a pro-inflammatory role of vitamin D has also been suggested(Reference Sun and Zemel729), epidemiological data show an association between vitamin D deficiency and increased risk of several inflammatory diseases including type 1 diabetes and atherosclerosis(Reference Borradale and Kimlin730). Vitamin D has several anti-inflammatory actions. It blunts the pro-inflammatory effects of AGE on endothelial cells, suggesting that it acts as an endogenous vascular protector counteracting the possible deleterious effects of AGE(Reference Talmor, Golan and Benchetrit731). Vitamin D inhibits the proliferation of lymphocytes and induces their apoptosis(Reference Martinesi, Treves and d'Albasio732). In addition, vitamin D affects the expression of ICAM-1 on mononuclear cells and on endothelial cells, suggesting that it suppresses the recruitment of leucocytes to sites of inflammation(Reference Martinesi, Treves and d'Albasio732). In vitro, vitamin D modulates the pro-inflammatory profile of monocytes/macrophages from type 2 diabetic patients(Reference Giulietti, van and Overbergh733). Vitamin D also suppresses TNF-α expression in monocytes/macrophages(Reference Cohen-Lahav, Douvdevani and Chaimovitz734) and down-regulates the expression of TLR2 and TLR4 in human monocytes(Reference Sadeghi, Wessner and Laggner735–Reference Do, Kwon and Park737). Indeed, vitamin D primes monocytes to respond less effectively to bacterial cell wall components, most probably due to the aforementioned suppression of TLR(Reference Sadeghi, Wessner and Laggner735). Vitamin D analogues selectively inhibit the inducible cyclo-oxygenase-2(Reference Aparna, Subhashini and Roy738), which could be viewed as an anti-inflammatory action. Exposure to vitamin D increases the rate of the de-phosphorylation of activated extracellular signal-regulated kinases(Reference Ziv, Rotem and Miodovnik739), a subset of the mammalian MAPK family involved in inflammatory processes. Serum vitamin D concentration was associated with leucocyte telomere length in 2160 women aged 18–79 years (mean age 49 years) and was inversely associated with CRP concentration(Reference Richards, Valdes and Gardner740). Within any tertile of vitamin D status, telomere length was longer in those with lower CRP concentrations, suggesting that inflammation plays a role in those processes associated with telomere shortening.
There are several studies looking at the association between vitamin D status (typically assessed as serum or plasma 25(OH)D3) and various markers of low-grade inflammation in different population subgroups. Shea et al. (Reference Shea, Booth and Massaro741) reported no significant association between plasma vitamin D concentration and concentrations of CRP, fibrinogen, TNF-α, IL-6, sTNFR2, sICAM-1, MCP-1, sP-selectin and sCD40 ligand in almost 1400 American adults with a mean age of 59 years. Likewise, there was no association of vitamin D status with serum CRP concentration in 650 Amish(Reference Michos, Streeten and Ryan742) or with CRP, sICAM-1, MCP-1, IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12, IL-13 or IL-17 concentrations in 437 overweight adults(Reference Jorde, Sneve and Torjesen743). Using data from over 6500 British adults aged 45 years, Hypponen et al. (Reference Hypponen, Berry and Cortina-Borja744) reported that the significant inverse associations between serum 25(OH)D3 concentration and serum CRP and fibrinogen were lost after adjustment for confounding factors. Among 261 healthy men and women, plasma 25(OH)D3 was not correlated with resistin, adiponectin or IL-18 concentrations but was inversely correlated with leptin concentration(Reference Vilarrasa, Vendrell and Maravall745). Among forty-four morbidly obese subjects, 25(OH)D3 did not correlate with leptin, resistin, adiponectin or IL-18. Jablonski et al. (Reference Jablonski, Chonchol and Pierce746) examined the inflammatory phenotype of endothelial cells collected from the antecubital vein of middle-aged and older subjects: endothelial cell expression of NF-κB and of IL-6 were both higher in vitamin D-deficient subjects, and IL-6 expression was inversely related to 25(OH)D3 concentration. Using data pooled from thirty-six healthy subjects, twenty-four type 1 diabetics and twenty-six type 1 diabetics with microvascular complications, Devaraj et al. (Reference Devaraj, Yun and Duncan-Staley747) found significant inverse relationships between serum 25(OH)D3 and CRP concentration, monocyte NF-κB activation and TLR4 expression. Most recently, NHANES data for 5867 adolescents aged 12–19 years showed no relationship between serum 25(OH)D3 and CRP concentrations(Reference Ganji, Zhang and Shaikh748). Thus, association studies have consistently found little, if any, association between vitamin D status and circulating markers of inflammation. However, two studies that investigated cellular markers of inflammation both reported an anti-inflammatory effect of vitamin D(Reference Devaraj, Yun and Duncan-Staley747, Reference Ganji, Zhang and Shaikh748).
Most intervention studies with vitamin D have failed to identify a reduction in markers of low-grade inflammation(Reference Hypponen, Berry and Cortina-Borja744, Reference Pittas, Harris and Stark749, Reference Tasinato, Boscoboinik and Bartoli750). These studies each had a different design. Pittas et al. (Reference Pittas, Harris and Stark749) provided 700 IU (17·5 μg) vitamin D3 plus 500 mg calcium citrate daily to adult non-diabetics for 3 years in a double-blind, randomised, controlled trial and found no effect on plasma CRP or IL-6. Witham et al. (Reference Witham, Crighton and Gillespie751) provided 100 000 IU (2500 μg) vitamin D2 or placebo to elderly patients with systolic heart failure at study entry (week 0) and after 10 weeks and found no effect on plasma TNF-α at 10 or 20 weeks. Jorde et al. (Reference Jorde, Sneve and Torjesen743) provided 40 000 or 20 000 IU (1000 or 500 μg) vitamin D3 per week or placebo to overweight subjects for 1 year; there was no difference between groups in the changes in concentrations of CRP, sICAM-1, MCP-1, IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12, IL-13 or IL-17 over the course of the study. Thus, these intervention studies suggest little anti-inflammatory action of vitamin D in the sorts of subjects studied. However, compared with placebo, vitamin D (3300 IU (82.5 μg)/d for 12 months) resulted in a decrease (by 10 %) in TNF-α concentration in overweight subjects on a weight-reduction programme(Reference Zittermann, Frisch and Berthold752). This finding suggests that vitamin D dose, the nature of the supplementation regimen and the health status of the individuals under study, as well as starting vitamin D status, may all be important factors in determining the effect of supplemental vitamin D.
Antioxidant vitamins (vitamin C, vitamin E and carotenoids)
Vitamin C is a potent water-soluble antioxidant. Ascorbate is the active form of vitamin C and exerts antioxidant function. Upon its action as an antioxidant, ascorbate is oxidised to dehydroascorbate which can be reduced back to ascorbate by the oxidation of reduced glutathione to glutathione disulfide. Ascorbate is present at high concentrations in leucocytes, suggesting a significant role in inflammation and in protection against oxidative damage. Patients with the metabolic syndrome or diabetes showed decreased vitamin C status and increased lipid peroxidation(Reference Palmieri, Grattagliano and Portincasa753–Reference Ford, Mokdad and Giles756). These findings could be explained by increased oxidative stress as a result of diabetes leading to consumption of ascorbate or a role of low vitamin C status as a risk factor for the development of diabetes. Obese subjects have lower plasma vitamin C concentrations than non-obese, and obesity was associated with moderately elevated CRP concentrations(Reference Aasheim, Hofso and Hjelmesaeth757). In another study, low plasma vitamin C concentrations were related to central fat distribution, independent of BMI(Reference Canoy, Wareham and Welch758).
Vitamin E is an umbrella term for a number of tocopherols and tocotrienols, although dietary vitamin E mainly consists of α- and γ-tocopherols. Vitamin E is a potent chain-breaking antioxidant that acts mainly in the lipid phase and interrupts the chain reaction of lipid peroxidation and, consequently, prevents the propagation of free radical-initiated reactions. There are differences in the antioxidant activity between α- and γ-tocopherols. In vitro, vitamin E exerts a range of anti-inflammatory actions with regard to the production of pro-inflammatory cytokines and eicosanoids and adhesive interactions of monocytes with endothelial cells(Reference Tasinato, Boscoboinik and Bartoli750, Reference Devaraj, Li and Jialal759–Reference Devaraj and Jialal761). There is growing evidence that γ-tocopherol, in contrast to α-tocopherol, exerts anti-inflammatory properties which can be explained by an unsubstituted position on the chromanol ring providing γ-tocopherol with the ability to trap reactive N species and subsequent formation of 5-nitro-γ-tocopherol(Reference Cooney, Franke and Harwood762, Reference Christen, Woodall and Shigenaga763). Supplementation with α-tocopherol decreases γ-tocopherol concentrations(Reference Handelman, Machlin and Fitch764, Reference Sundl, Resch and Bergmann765) due to a preference of the α-tocopherol transfer protein in the liver for α-tocopherol. This results in increased metabolism to carboxyethyl-hydroxychroman derivatives and excretion of the metabolites in the urine(Reference Lodge, Ridlington and Leonard766). γ-Tocopherol and γ-carboxyethyl-hydroxychroman exert actions that are not shared by α-tocopherol and α-carboxyethyl-hydroxychroman(Reference Jiang, Christen and Shigenaga767).
Carotenoids include, among others, α-carotene, β-carotene, lycopene, β-cryptoxanthin, lutein and zeaxanthin. They are highly prevalent in red, yellow and green vegetables and fruits. Carotenoids exert antioxidant properties and some of them serve as provitamin A. Among 3258 healthy men (age 68 (SD 5) years; BMI 26·7 (SD 3·6) kg/m2), dietary vitamin C intake and plasma vitamin C concentrations were both inversely associated with CRP concentrations(Reference Wannamethee, Lowe and Rumley329). Among 5181 subjects from the MESA aged 45–84 years, intakes of vitamin C, vitamin E or β-carotene were not associated with CRP, IL-6 or fibrinogen concentrations after adjusting for a number of factors(Reference De Oliveira, Alonso and Lee768). Recent data from the Womens’ Health Study reported that higher plasma concentrations of α- and β-carotene were associated with lower plasma CRP concentrations(Reference Wang, Gaziano and Norkus769). Similar results were reported from NHANES III for carotenoids and CRP(Reference Kritchevsky, Bush and Pahor770, Reference Erlinger, Guallar and Miller771). However, the use of latex-enhanced nephelometry instead of a high-sensitivity ELISA, and the fact that 74 % of the population had levels below the assay's detection limit, limits the interpretation of their results. In another study, dietary intakes of vitamin C, vitamin E and β-carotene did not predict the concentrations of CRP, IL-6 or TNF-α in normal-weight, overweight and obese Swiss children aged 6–14 years, but they were significant predictors of leptin concentrations(Reference Aeberli, Molinari and Spinas772). Using data from a small number of middle-aged (mean age 44 years) vegetarians and omnivores (n 30 per group), plasma vitamin C concentrations were found to be inversely associated with CRP concentrations(Reference Szeto, Kwok and Benzie295). In 379 Dutch adults, serum lutein and lycopene concentrations were inversely associated with sICAM-1 concentrations, serum β-carotene with total blood leucocyte numbers and CRP concentrations, serum vitamin C with CRP concentrations, while plasma α-tocopherol concentrations were positively associated with CRP concentrations(Reference van Herpen-Broekmans, Klopping-Ketelaars and Bots773). Among 437 Japanese subjects, serum β-carotene concentrations were positively associated with adiponectin concentrations(Reference Suzuki, Inoue and Hashimoto774). Other associations among α- and β-carotene and CRP and IL-6 concentrations were lost after adjustment for confounding factors. A larger Japanese study (n 778 men and 1404 women) reported an inverse association between serum vitamin C and CRP concentrations after adjustment for confounders(Reference Kubota, Moriyama and Yamagishi775). The association was strongest in non-smokers, in non-overweight women and in postmenopausal women. Recently, plasma vitamin C concentrations have been reported to be inversely associated with plasma CRP concentrations in a small number of lean and obese men (n 8 per group) with a mean age of 21 years(Reference Mah, Matos and Kawiecki776). In patients with coronary artery disease, plasma concentrations of β-carotene, but not lycopene, were inversely correlated with plasma CRP concentrations(Reference Jonasson, Wikby and Olsson777). In one prospective study with young normal-weight adults, a high intake of carotenoids (α-carotene, β-carotene, zeaxanthin/lutein and β-cryptoxanthin) was inversely related to plasma CRP and ICAM concentrations after 7 and 15 years of follow-up(Reference Hozawa, Jacobs and Steffes778). In a second prospective study, a high intake of carotenoids in elderly subjects (BMI 28·8 (SD 6·8) kg/m2) was associated with lower plasma IL-6 concentrations, while α-tocopherol did not correlate significantly(Reference Walston, Xue and Semba779). Among 704 70-year-old Swedish men, dietary intakes of vitamin C and α-tocopherol, but not of β-carotene, were inversely associated with CRP and IL-6 concentrations measured 7 years after the dietary information was collected(Reference Helmersson, Arnlov and Larsson780). Thus, overall, cross-sectional and prospective studies fairly consistently demonstrate that a higher intake and status of vitamin C, vitamin E and carotenoids is associated with lower levels of low-grade inflammation.
Taking 1 g of vitamin C or 533 mg of α-tocopherol before consuming a high-fat test meal blunted the acute CRP response to the meal(Reference Carroll and Schade781). Plasma 8-iso PGF2α and MCP-1 concentrations decreased after consumption of 72 mg/d of vitamin C from a vegetable soup(Reference Madsen, Schmidt and Christensen634); concentrations of TNF-α, IL-1β and IL-6 did not change. Vitamin C (1 g/d for 14 d) did not alter sICAM-1 concentrations or markers of monocyte and neutrophil activation (neopterin and elastase, respectively) in smokers or non-smokers (n 20 of each)(Reference Scott, Poston and Wilson782). Likewise, vitamin C supplements (250 mg three times per week for 2 months) did not alter plasma CRP concentrations in thirty-three chronic haemodialysis patients(Reference Fumeron, Nguyen-Khoa and Saltiel783). Tomato juice (delivering 20·6 mg lycopene/d) was compared with tomato juice fortified with vitamin C (delivering 435 mg vitamin C/d) over a 2-week intervention period: the vitamin C-enriched juice did not affect plasma CRP, TNF-α or IL-1β concentrations in healthy volunteers(Reference Jacob, Periago and Bohm784). Vitamin C (1 g/d for 6 months) lowered sP-selectin, but not sVCAM-1, sICAM-1 or sE-selectin concentrations in patients with chronic degenerative aortic stenosis(Reference Tahir, Foley and Pate785); combining vitamin C with α-tocopherol (267 mg/d) lowered sICAM-1 concentrations. Smokers given 515 mg vitamin C/d for 2 months had a 24 % reduction in plasma CRP concentrations(Reference Block, Jensen and Dietrich786). A mixture of vitamin C with α-tocopherol (371 mg/d), γ-tocopherol (171 mg/d), mixed tocotrienols (252 mg/d) and α-lipoic acid (95 mg/d) resulted in a smaller effect (4·7 % reduction that was not significant), suggesting that tocopherols, tocotrienols or α-lipoic acid, or the combination, prevents the anti-inflammatory effect of vitamin C. Vitamin C (2 g/d for 4 weeks) did not affect serum IL-6, IL-1b, sVCAM-1 or sICAM-1 concentrations in smokers, but combining 533 mg/d α-tocopherol with the vitamin C decreased the concentrations of all four inflammatory mediators; the combination of vitamin C and 267 mg/d α-tocopherol was without effect(Reference Tousoulis, Antoniades and Tentolouris787).
α-Tocopherol (800 mg/d for 3 months) decreased plasma CRP concentrations in type 2 diabetics(Reference Devaraj and Jialal788). Dalgard et al. (Reference Dalgard, Nielsen and Morrow789) examined the effect of a 28 d intervention with orange and blackcurrant juice (500 ml/d), vitamin E (15 mg RRR-α-tocopherol/d) or the combination of the two in patients with peripheral vascular disease. The juice but not vitamin E decreased CRP (by 11 %) and fibrinogen, but there was no effect on IL-6 or PAI-1. In a randomised, placebo-controlled, double-blind trial, subjects with the metabolic syndrome received 800 mg/d α-tocopherol, 800 mg/d γ-tocopherol, a combination of both (800 mg/d each) or placebo; there was a decrease in CRP concentrations only in the combined α- and γ-tocopherol supplementation group, while TNF-α decreased with α-tocopherol alone or in combination with γ-tocopherol(Reference Devaraj, Leonard and Traber790). Biomarkers of oxidative stress decreased with α-tocopherol, γ-tocopherol or the combination, while nitrotyrosine, a biomarker of nitrative stress, decreased with γ-tocopherol alone or in combination with α-tocopherol(Reference Yusof, Miles and Calder646–Reference Rossi, Lombardo and Lacorte648). In another randomised, placebo-controlled, double-blind trial, overweight and obese and normal-weight young adults completed a standardised 30 min cycle exercise bout before and after 8 weeks of supplementation with 800 IU/d of vitamin E (form not stated), 500 mg/d of vitamin C and 10 mg/d of β-carotene(Reference Vincent, Bourguignon and Vincent791). Adiponectin concentrations were increased by 22 % in the overweight and by 3 % in the normal-weight group receiving the supplement, but was reduced in the placebo group. Changes in circulating IL-6 concentrations during exercise were lower in the supplemented groups, as were the changes in lipid hydroperoxides(Reference Vincent, Bourguignon and Vincent791). In contrast, a double-blind, placebo-controlled trial, in which type 2 diabetics received 500 mg/d α-tocopherol or mixed tocopherols containing 315 mg/d γ-tocopherol, reported no effect on CRP, IL-6, TNF-α or MCP-1 concentrations, although there was a decrease in plasma F2-isoprostane concentrations, a biomarker of lipid peroxidation, in the mixed tocopherol group(Reference Wu, Ward and Indrawan792). A small study conducted in healthy subjects (n 12) and patients with CHD (n 12) investigated the effect of increasing intake of α-tocopherol (100, 200 and 400 mg/d each for 3 weeks sequentially)(Reference Leichtle, Teupser and Thiery793). At 200 mg/d, plasma CRP, IL-6 and fibrinogen concentrations were decreased in the CHD patients. A long-term (3 years) intervention with the combination of vitamin C (500 mg/d) and α-tocopherol (182 mg/d) in 45–69-year-old men did not alter CRP, TNF-α or IL-6 concentrations(Reference Bruunsgaard, Poulsen and Pedersen794). In one study, fifteen children with familial hyperlipidaemia given vitamins C plus E (500 mg/d and 400 IU/d, respectively) for 6 weeks showed no changes in CRP concentration(Reference Engler, Engler and Malloy795). In another study, six weeks’ supplementation with vitamin C (1 g/d) plus vitamin E (300 mg/d RRR-α-tocopherol) did not alter the magnitude of the increase in circulating CRP, TNF-α and IL-6 concentrations seen after running a marathon(Reference Mastaloudis, Morrow and Hopkins796). Older men given 1 g/d of vitamin C plus 1000 mg/d vitamin E for 4 weeks showed a decrease in plasma TNF-α concentrations(Reference Rizzo, Abbatecola and Barbieri797). Recently, a mixture of fruit-derived antioxidants has been found not to alter CRP or IL-6 concentrations over 12 weeks in type 2 diabetics(Reference Rytter, Vessby and Asgard798). In an intervention study with carotenoid-rich vegetables and fruits, plasma concentrations of α- and β-carotene, but not other carotenoids, were inversely associated with plasma CRP concentrations in healthy normal-weight men(Reference Watzl, Kulling and Moseneder345). Lycopene (80 mg/d for 1 week) did not affect plasma CRP, sVCAM-1 or sICAM-1 concentrations in men and women with a mean age of 23 years(Reference Denniss, Haffner and Kroetsch799).
Thus, in contrast to observational studies that provide a fairly consistent picture of an anti-inflammatory effect of vitamin C, vitamin E and carotenoids, intervention studies using supplements of these antioxidant vitamins either alone or in various combinations provide a less consistent set of observations. A number of studies do demonstrate a reduction in the concentrations of circulating inflammatory markers in a variety of subgroups of individuals, including the overweight persons and diabetics, but quite a number of studies did not find an effect. The lack of consistency may be related to differences in: dose of antioxidant used (however, typically the doses used are much greater than those that can be readily achieved in the diet and are therefore much greater than would have been present in the diets of subjects investigated in the observational studies); duration of treatment (typically a few weeks to a few months); sample size which has often been small; characteristics of the populations studied (e.g. age, healthy v. diseased, type of disease, smokers v. non-smokers); background diet; interactions among the different antioxidant vitamins used, since they may have different anti-inflammatory potencies and some may even act in a pro-inflammatory way under certain conditions; degree of oxidative stress present. One other factor that has recently been identified is genetic differences among individuals, which may have an impact on the ability of antioxidants to exert an anti-inflammatory effect. Such an effect has been identified by Belisle et al. (Reference Belisle, Leka and Delgado-Lista800) who showed that the ability of α-tocopherol to lower the LPS-stimulated production of TNF-α by whole blood was determined in part by polymorphisms within the TNF-α gene. Thus, it is possible that a greater or lesser anti-inflammatory effect of antioxidant vitamins will be observed in people with different genotypes related to inflammatory processes. Clearly, this needs greater exploration in properly designed studies.
Flavonoids
Polyphenols are secondary metabolites of plants involved in pigmentation, reproduction and protection against pathogens. There are more than 8000 known polyphenolic substances sharing a common chemical structure (hydroxyl group on an aromatic ring) with different constituents. Flavonoids are the most abundant polyphenols present in the human diet, and they can be divided into several classes according to different constituents such as flavanones, flavones, flavanols and flavonols. They can be found in almost all plant foods and, among the flavonols, myricetin, kaempferol and quercetin are the most representative, while catechins are the most abundant flavanols contained in tea leaves. Flavanones are mainly represented in the diet by taxifolin, naringinin and hesperitin. The main sources of flavanones are citrus fruits. Flavones are less common. In addition to these, other classes of flavonoids are present in the diet such as proanthocyanidins and their oligomers.
The intake of flavonols and flavones was not correlated with plasma concentrations of CRP or IL-6 in healthy women (BMI 25·8–26·2 kg/m2)(Reference Song, Manson and Buring801). While this study used a rather limited flavonoid database, a more recent cross-sectional study applying a comprehensive flavonoid database reported anti-inflammatory effects associated with a high flavonoid intake: total flavonoid, flavonol and anthocyanidin intakes were inversely associated with plasma CRP concentrations(Reference Chun, Chung and Claycombe323). Data from the Nurses’ Health study were used to assess the relationship between flavonoid intake and biomarkers of inflammation(Reference Landberg, Sun and Rimm802): intake of six flavonoid subclasses (flavonols, flavones, flavanones, flavan-3-ols, anthocyanidins and polymeric flavonoids) was assessed using a FFQ administered in 1990 and blood samples collected in 1989–90 were used to measure concentrations of CRP, IL-6, IL-18, sTNFR2, sVCAM-1 and sE-selectin. Multivariate-adjusted mean plasma IL-18 concentrations were lower (by 9, 11 and 8 %, respectively) for women in the highest intake quintile of flavones, flavanones and total flavonoids compared with those in the lowest quintiles. Multivariate-adjusted geometric plasma sVCAM-1 concentrations were lower by 4 % in women in the highest intake quintile of flavonol compared with those in the lowest quintile. Thus, the study suggests that higher intakes of selected flavonoid subclasses are associated with modestly lower concentrations of some inflammatory biomarkers.
In a randomised human intervention trial with healthy normal-weight adults, supplementation with a bilberry extract providing 300 mg anthocyanins/d (equal to 100 g of fresh bilberries) reduced plasma concentrations of several NF-κB-induced pro-inflammatory cytokines (IL-8, RANTES and IFN-α)(Reference Karlsen, Retterstol and Laake337). In addition, NF-κB-inducing cytokines (IL-4 and IL-13) tended to differ from controls, while plasma CRP concentrations did not change(Reference Karlsen, Retterstol and Laake337). In subjects who had survived myocardial infarction and had received statin therapy for at least 6 months, supplementation with a chokeberry flavonoid extract for 6 weeks significantly decreased CRP and MCP-1 concentrations, while adiponectin was significantly increased(Reference Naruszewicz, Laniewska and Millo803). In contrast, although the consumption of black tea resulted in increased plasma catechin concentrations, these were not associated with changes in plasma CRP concentrations in healthy overweight and obese adults(Reference Widlansky, Duffy and Hamburg389). A similar negative effect on CRP was observed in a double-blind, placebo-controlled, cross-over study with healthy adults (mean BMI 25·8 kg/m2) who were supplemented with a sea buckthorn flavonol extract during 4 weeks(Reference Suomela, Ahotupa and Yang804). Quercetin (50, 100 or 150 mg/d for 2 weeks) did not affect serum concentrations of TNF-α in healthy adults(Reference Egert, Wolffram and Bosy-Westphal805). Overweight or obese subjects aged 25–65 years with metabolic syndrome traits received 150 mg quercetin/d in a double-blind, placebo-controlled, cross-over trial with 6-week treatment periods separated by a 5-week washout period: quercetin did not affect CRP or TNF-α concentrations compared with placebo(Reference Egert, Bosy-Westphal and Seiberl806). Quercetin (1 g/d for 21 d) failed to attenuate muscle inflammation in ultramarathon runners(Reference Nieman, Henson and Davis807) and in trained cyclists(Reference Nieman, Henson and Davis808, Reference McAnulty, McAnulty and Nieman809). However, leucocyte IL-8 and IL-10 mRNA were significantly reduced, indicating that a high dose of quercetin may target blood cells but not the muscle tissue.
Phyto-oestrogens
Genistein is an isoflavone and a phyto-oestrogen which primarily occurs in soyabeans. Native phyto-oestrogens exist as glycosides, while in experimental studies, mostly the aglycones have been used. In two intervention studies with healthy postmenopausal women, the intake of genistein (54 or 40 mg/d) for 6 months did not significantly affect plasma CRP concentrations(Reference D'Anna, Baviera and Corrado810, Reference Yildiz, Kumru and Godekmerdan811). The intake of soya either high or low in isoflavones for 1, 2 or 4 months had also no effect on CRP, SAA or TNF-α concentrations in hypercholesterolaemic men or in postmenopausal women(Reference Jenkins, Kendall and Jackson356, Reference McVeigh, Dillingham and Lampe359, Reference Ryan-Borchers, Park and Chew362). In obese postmenopausal women, the combination of exercise with a soya isoflavone supplement (duration 6 months) did not decrease plasma CRP concentration compared with exercise+placebo(Reference Aubertin-Leheudre, Lord and Khalil812). Consumption of a soya isoflavone-enriched cereal bar (50 mg/d) for 8 weeks by postmenopausal women had no effect on CRP or other plasma markers of inflammation(Reference Hall, Vafeiadou and Hallund813). A higher intake of soya isoflavones (114 mg/d) for 3 months also did not reduce serum CRP or sE-selectin concentrations in postmenopausal women(Reference Nikander, Metsa-Heikkila and Tiitinen814). In another study, isoflavone-rich soya (107 mg/d as aglycone; 50 % as genestein) for 6 weeks did not affect sVCAM-1, sICAM-1 or sE-selectin concentrations in healthy postmenopausal women compared with isoflavone-poor soya(Reference Steinberg, Guthrie and Villablanca815). In contrast, a randomised, controlled study providing pasta naturally enriched with isoflavone aglycones (33 mg/d) to overweight hypercholesterolaemic subjects reported significantly reduced plasma CRP concentrations, which returned to baseline when subjects were switched to conventional pasta(Reference Clerici, Setchell and Battezzati816). Overall, the majority of the studies with soya-derived isoflavones did not observe a significant effect on inflammatory processes in human subjects. In contrast, a lignan complex (500 mg/d) isolated from flax given to healthy postmenopausal women significantly reduced plasma CRP concentration during the 6-week placebo-controlled intervention. No significant differences were found for IL-6, TNF-α, sICAM-1, sVCAM-1 or MCP-1 concentrations(Reference Hallund, Tetens and Bugel817). In a recent study, flaxseed-derived lignans (360 mg/d) lowered CRP concentrations in type 2 diabetic women but not in men over 12 weeks compared with placebo, with no effect on IL-6 concentrations(Reference Pan, Demark-Wahnefried and Ye818). A cross-sectional study in 242 men and postmenopausal women in Northern Italy revealed inverse associations between dietary intake of the lignans matairesinol and secoisolariciresinol and plasma concentrations of sICAM-1(Reference Pellegrini, Valtuena and Ardigo819).
Other factors
Gut microbiota and probiotics
Probiotics are ‘live micro-organisms which, when consumed in adequate quantities, confer a health benefit on the host’(Reference Salminen, Bouley and Boutron-Ruault820, Reference Nova, Warnberg and Gomez-Martinez821). One differential characteristic of probiotics compared with other micro-organisms is their ability to survive during gastrointestinal transit(Reference Ljungh and Wadstrom822). This allows them to interact with commensal microbiota and/or intestinal epithelial cells and also with gut-associated lymphoid cells, which results in the induction or modulation of a number of biological activities that can provide beneficial effects for health. The capacity of probiotics to modulate the mucosal immune system is regarded as one of the most obvious beneficial properties. The microbiota composition and the related local intestinal metabolism undergoes significant changes in various disease states that are characterised by chronic inflammation, such as inflammatory bowel disease(Reference Sartor823), colorectal cancer and obesity(Reference Ley, Backhed and Turnbaugh824, Reference Tennyson and Friedman825). Mice raised under germ-free conditions fail to develop experimental colitis, suggesting an important modulating role of intestinal microbiota on local inflammatory processes(Reference Rescigno826). Thus, there seems to be a strong association between the nature of the gut microbiota and inflammation.
A number of studies have investigated the effects of various probiotics on aspects of inflammation in a variety of subject and patient groups, as reviewed elsewhere(Reference Lomax and Calder827). However, relatively few studies have focused on circulating markers of inflammation in persons without an established inflammatory disease. In a small study in institutionalised elderly subjects (mean age 76 years), there was no effect of the combination of Bifidobacterium longum and Lactobacillus acidophilus (8 × 109 colony forming units (CFU) of each/d for 28 d) on serum TNF-α concentration(Reference De Simone, Ciardi and Grassi828). L. salivarius UCC118 (1010 CFU/d for 21 d) did not affect serum IL-1α, IL-1β, IL-4, sIL-2R, sIL-6R, TNF-α or IFN-γ concentrations in healthy adults aged 20–65 years(Reference Collins, Dunne and Murphy829). Likewise, the combination of L. gasseri CECT5714 and L. coryniformis CECT5711 (2 × 109 CFU of each/d) with Staphylococcus thermophilus (108 CFU/d) for 2 or 4 weeks had no effect on serum TNF-α or IL-12 concentrations in healthy adults aged 23–43 years(Reference Olivares, Díaz-Ropero and Gomez830, Reference Olivares, Paz Diaz-Ropero and Gomez831). Kekkonen et al. (Reference Kekkonen, Lummela and Karjalainen832) compared L. rhamnosus GG ATCC53103 (1·6 × 1010 CFU/d) with Bifidobacterium animalis ssp. lactis Bb12 (3·5 × 1010 CFU/d) and Propionibacterium freudenreichii ssp. Shermani JS (3·3 × 1010 CFU/d) for 3 weeks in healthy subjects with a mean age of 44 years (range 23–58 years). There was no effect of serum TNF-α, IL-6, IL-10 or IFN-γ concentrations, but serum CRP concentration decreased in the L. rhamnosus group. Ouwehand et al. (Reference Ouwehand, Tiihonen and Saarinen833) conducted a 6-month placebo-controlled trial with B. animalis ssp. lactis Bb12 (109 CFU/d) or the combination of B. longum 2C and B. longum 46 (109 CFU of each/d) in institutionalised elderly subjects (mean age 84 years). Although there were some changes over time, the groups did not differ in serum TNF-α, IL-10 or TGF-β concentrations. Thus, although there is the potential for probiotics to lower markers of chronic low-grade inflammation, intervention studies performed to date in human volunteers rarely demonstrate this effect.
Prebiotics
There are two recent definitions of prebiotics: ‘a selectively fermented ingredient that allows specific changes, both in the composition and/or activity in the gastrointestinal microflora that confers benefits upon host well-being and health’(Reference Gibson, Probert and Loo834), and ‘a non-viable food component that confers a health benefit on the host associated with modulation of the microbiota’(835). Typically, though not exclusively, prebiotics are carbohydrates including inulin-type fructans (including oligofructose, fructo-oligosaccharides), lactulose, galacto-oligosaccharides, xylo-oligosaccharides, d-tagatose, resistant starch, soyabean oligosaccharides, pectin, guar, carrageenan, konjac glucomannans, alginates and β-glucans from oat, barley and mushroom. Prebiotics escape digestion in the upper gastrointestinal tract and reach the large intestine virtually intact, where they are fermented by the microbiota and express their prebiotic activity(Reference Gibson, Probert and Loo834). The latter is most probably mediated through a quantitative increase in commensal bacteria (e.g. bifidobacteria and lactobacilli), which interact with other members of the gut microbiota. Further, changes in the microbiota enzyme activities, leading to a reduction of unfavourable substances that have an impact on disease risk, such as secondary bile acids, and the production of metabolites such as SCFA and vitamins also contribute to their impact on health and disease(Reference Gibson, Probert and Loo834). In high-fat-fed mice, oligofructose restored gut bifidobacteria, and normalised systemic endotoxin levels and the inflammatory state(Reference Cani, Neyrinck and Fava836). Institutionalised elderly individuals (mean age 85 years) supplemented with oligofructose (8 g/d for 3 weeks) showed increased faecal bifidobacteria counts and decreased expression of IL-6 mRNA in blood monocytes(Reference Guigoz, Rochat and Perruisseau-Carrier837). In another study, in poorly nourished elderly subjects (age >70 years), oligofructose (1·95–3·9 g/d for 12 weeks as part of a nutritional supplement) had no effect on plasma TNF-α or sIL-6R concentrations(Reference Schiffrin, Thomas and Kumar838). Prebiotics have been studied in the context of inflammatory conditions, especially those involving the gastrointestinal tract, as reviewed elsewhere(Reference Lomax and Calder839). There is some evidence of beneficial effects but results are inconsistent. There are few studies of prebiotics and circulating inflammatory markers in the context of chronic low-grade inflammation. Thus, it is premature to draw conclusions about this relationship.
Hydration
When fluids are consumed, water distributes between intra- and extracellular compartments according to osmotic load. When water enters the cells, they swell and when water is lost from the cells, for example, during dehydration, the cells will shrink. The maintenance of adequate cell volume can have a profound effect on protein function and cellular performance. Cells employ an array of mechanisms to maintain cell volume constancy, including altered transport across the cell membrane and metabolism. Hormones and mediators may modify the activity of these cell volume regulatory mechanisms and thus influence cell volume-sensitive functions. Cell volume regulatory mechanisms, therefore, participate in the signalling of those hormones and mediators(Reference Haussinger and Lang840). Many metabolic pathways are sensitive to cell volume(Reference Lang, Busch and Ritter841), as a result of activation, inhibition or altered expression of enzymes. Cellular volume has the potential to have an impact directly or indirectly on inflammation, but the effect of hydration status on low-grade inflammation is not well documented.
Summary, conclusions and research gaps
Inflammation is part of the normal host defence mechanism against infections. However, inflammatory mediators and the inflammatory response can be damaging to the host if not regulated appropriately and numerous diseases and conditions have an overt chronic inflammatory basis. It is now recognised that a lower level of inflammation, here termed chronic low-grade inflammation, can also persist and may be a cause of, or result from, the obese state. Adipose tissue releases many of the characteristic mediators of inflammation including some of the classic pro-inflammatory cytokines and chemokines, as well as adiponectin which is considered to be anti-inflammatory. The source of these mediators within adipose tissue is not clear, but infiltrating macrophages seem to be especially important in this regard, although adipocytes themselves express some of the inflammatory mediators. Obese people have higher circulating concentrations of many inflammatory markers (measured in the ‘resting’ state, i.e. in fasted blood), and these are believed to play a role in causing insulin resistance and in other metabolic disturbances of the metabolic syndrome and type 2 diabetes. Blood concentrations of inflammatory markers are lowered following weight loss, whether this is induced by diet or surgery, which most probably reflects the decrease in adipose tissue mass. In the hours following the consumption of a meal, there is an elevation in the concentrations of several inflammatory mediators in the bloodstream. This postprandial inflammatory response is greater in obese subjects and type 2 diabetics. Both high-glucose and high-fat meals induce postprandial inflammation, and it is exaggerated by a high meal content of AGE and partly ablated by the inclusion of certain antioxidants or antioxidant-containing foods within the meal. These latter observations link postprandial inflammation to oxidative stress. Physical activity decreases low-grade inflammation. Exercise itself is associated with transient and local inflammation (e.g. in muscle) that may, in fact, be important in inducing a protective and ultimately healthy anti-inflammatory response. In addition to the direct effect of foods and their constituents on postprandial inflammation, diet has an impact on chronic low-grade inflammation, manifested as the basal (i.e. fasting state) concentrations of inflammatory markers in the bloodstream, including cytokines, chemokines, acute-phase proteins, soluble adhesion molecules and cytokine receptors, etc. Effects of diet and dietary components on low-grade inflammation have been identified through cross-sectional, prospective and intervention studies. The former two study designs frequently involve large numbers of subjects, while the intervention studies often have a small sample size and a limited duration which might limit their ability to identify effects; compliance may also be a limitation of intervention studies. Healthy eating patterns such as the Mediterranean diet, vegetarian diets and adherence to the Food Guide Pyramid are associated with lower concentrations of inflammatory markers; this is observed mainly from cross-sectional and prospective studies, although intervention studies with the Mediterranean diet have been positive. Among the components of healthy diets, whole grains, vegetables and fruits, and fish are all seen to be associated with lower inflammation. Strong evidence in favour of an anti-inflammatory effect of tea (black or green), coffee (caffeinated or decaffeinated) and cocoa is lacking, despite positive effects on oxidative stress and the anti-inflammatory effects, mainly demonstrated in model systems (e.g. cell cultures), of components of these foods. Alcohol appears to have a ‘U-shaped’ effect on low-grade inflammation, with the most protective action (i.e. the lowest inflammatory marker concentrations) corresponding to one or two alcoholic drinks per d. Heated meals high in AGE and ALE enhance oxidative stress and inflammation; intervention studies with low- and high-AGE meals either acutely or chronically have associated AGE with increased inflammatory marker concentrations and show that these are decreased by meals low in AGE. However, AGE and ALE are also generated in vivo and also as a result of other, non-food-related, environmental exposures (e.g. smoking), and so the overall impact of foods on the burden of AGE and ALE is not currently clear; this is compounded by technical difficulties in measuring these complex chemical entities in foods and in body fluids. Dietary fatty acids also influence low-grade inflammation; best studied are PUFA. Available data indicate that SFA and trans-MUFA are pro-inflammatory, and that one isomer of conjugated linoleic acid may also be. Relative to SFA, PUFA are anti-inflammatory. Marine n-3 PUFA have the greatest anti-inflammatory potential. Hyperglycaemia induces both postprandial and chronic low-grade inflammation, acting in part through oxidative stress. Dietary fibre decreases low-grade inflammation. There may also be a role for milk peptides, but these have not been sufficiently evaluated. Vitamin D has the potential to reduce low-grade inflammation, with plausible mechanistic actions demonstrated in model systems. There is good evidence from both model systems and from human observational and intervention studies that vitamin C, vitamin E and carotenoids decrease the concentrations of inflammatory markers. α- and γ-Tocopherols and the different carotenoids may have different anti-inflammatory properties and potencies. The majority of available evidence indicates that soya phyto-oestrogens and soya protein do not affect low-grade inflammation. There are likely to be many plant-derived substances that influence low-grade inflammation and which may have a role as part of a healthy ‘anti-inflammatory diet’. Several studies of various probiotic bacteria have failed to demonstrate any consistent effects on markers of chronic low-grade inflammation, despite their apparent effectiveness in higher-grade inflammatory states. The effect of prebiotics on chronic low-grade inflammation is not clear and is underexplored.
The key conclusions are as follows:
(1) A state of chronic low-grade inflammation exists and this is exaggerated in obese and type 2 diabetic individuals.
(2) Adipose tissue plays a role in establishing the chronic low-grade inflammatory state because cells within that tissue can produce and release the mediators involved.
(3) Chronic low-grade inflammation is believed to increase the risk of insulin resistance, type 2 diabetes and CVD.
(4) Following consumption of a meal, there is a transient state of inflammation that is linked with oxidative stress.
(5) Hyperglycaemia or a high-fat meal promote postprandial inflammation.
(6) A healthy diet is associated with decreased low-grade inflammation.
(7) Important protective factors in the diet are whole grains, fibre, vegetables, fruits, fish, PUFA, especially marine n-3 PUFA, vitamin C, vitamin E and carotenoids.
(8) Plant-derived flavonoids are likely to be protective.
(9) Moderate alcohol consumption decreases low-grade inflammation.
(10) Dietary factors that promote inflammation are oxidised lipids, SFA and trans-fatty acids.
(11) Underexplored dietary factors include milk peptides, vitamin D, probiotics and prebiotics.
A number of important research gaps were identified. Perhaps the most important is that there is no consensus regarding the inflammatory mediators which best represent chronic low-grade inflammation. Most studies have measured CRP, perhaps because it is long established, is linked to the risk of CVD and is routinely measured in clinical laboratories. However, it is not clear whether CRP is a ‘better’ marker of low-grade inflammation than any of the other mediators measured, which include a variety of cytokines, soluble cytokine receptors, chemokines, soluble adhesion molecules and so on. Furthermore, there has been little emphasis on anti-inflammatory mediators. An expert review of the area of inflammatory markers addressing the most valid and robust markers and including a consideration of anti-inflammatory factors is warranted. Identification of dietary components that promote or prevent postprandial inflammation and the underlying mechanisms involved is a further gap. The impact of many dietary components on chronic low-grade inflammation is underexplored in human intervention studies; such components include flavonoids and other phytochemicals, milk peptides, vitamin D, probiotics and prebiotics. Further, even where dietary components have been fairly well explored through intervention studies, many of the studies have been small in size and they have reported on different inflammatory outcomes at different time points. For these components (whole grains, fibre, vegetables, fruits, fish, PUFA, especially marine n-3 PUFA, vitamin C, vitamin E and carotenoids), a better knowledge of the dose–response effect, the threshold dose (if any) and the time required for an effect to occur would all be valuable information. Such variations in study design may also explain the discordant findings of studies with tea, coffee and cocoa, and these dietary components require further investigation in the context of robust markers of inflammation, appropriate sample size and duration of exposure, and exploration of dose–response relationships. Oxidative stress and inflammation are strongly interlinked and effects of dietary components appear to frequently involve increased or decreased oxidative stress. In this regard, modified food components such as AGE and ALE may be very potent markers of oxidative and inflammatory stress, although their causal impact remains elusive. However, further exploration and greater understanding of this area is impeded by lack of agreed analytical methods for quantifying AGE in foods and in biological fluids, with inter-laboratory cross-validation, and by lack of discrimination between protein-bound and free AGE. Finally, the emerging area of the role of gene polymorphisms in influencing or even determining the effect of nutrients on markers of low-grade inflammation requires much greater exploration. Until these gaps are filled, our understanding of the interaction between diet and postprandial and chronic low-grade inflammation will remain incomplete.
Acknowledgements
This work was commissioned by the Metabolic Syndrome and Diabetes Task Force of the European Branch of the International Life Sciences Institute (ILSI Europe). Industry members of this task force are Ajinomoto Europe, Coca-Cola Europe, Danisco, Danone, Kellog Europe, Kraft Foods Europe and Schwabegroup. For further information about ILSI Europe, please email info@ilsieurope.be or call +32 2 771 00 14. The opinions expressed herein and the conclusions of this publication are those of the authors and do not necessarily represent the views of ILSI Europe nor those of its member companies. Conflict of interest statement: P. C. C. has research funding from Beneo-Orafti, Beghin-Meiji, Abbott Nutrition and Vifor Pharma and is a consultant to Danone. F. B. was an employee of Cargill. T. B. was and J. O. is an employee of Nestlé. K. Cu. is an employee of Coca-Cola Europe. L. S. J. is an employee of ILSI Europe. M. L. is an employee of Danone. N. M. is an employee of Kraft Foods Europe. H. N. is an employee of Ajinomoto Europe. P. C. C., N. A., F. B., K. Cl., K. E., H. K., As. M., An. M., G. P., S. R., C. S., J. T., J. W. and B. M. W-R received an honorarium from ILSI Europe for their participation in this publication and/or reimbursement of their travel and accommodation costs for attending the related meetings. The remaining authors have no conflicts of interest. All authors contributed to discussions and had input into writing the article. P. C. C. and L. S. J. had responsibility for producing the final version of the article.
























