


EVIDENCE OF MICROGLIAL ACTIVATION AND BRAIN INFLAMMATION IN AUTISM
A study by Johns Hopkins University School of Medicine found evidence of microglial activation in individuals with autism or autism spectrum disorder (ASD) (Pardo et al., Reference Pardo, Vargas and Zimmerman2005). Indeed, several studies now provide evidence that children with autism suffer from an ongoing neuroinflammatory process in different regions of the brain involving microglial activation (Enstrom et al., Reference Enstrom, Lit, Onore, Gregg, Hansen and Pessah2005; Pardo et al., Reference Pardo, Vargas and Zimmerman2005; Vargas et al., Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005; Zimmerman et al., Reference Zimmerman, Jyonouchi, Comi, Connors, Milstein and Varsou2005). Evidence of neuroinflammation includes activated microglia and astrocytes found in post-mortem brain tissue (Pardo et al., Reference Pardo, Vargas and Zimmerman2005; Vargas et al., Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005; Morgan et al., Reference Morgan, Chana, Pardo, Achim, Semendeferi and Buckwalter2010) and irregular, proinflammatory cytokine profiles in the brain and cerebrospinal fluid of children with ASD (Vargas et al., Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005; Zimmerman et al., Reference Zimmerman, Jyonouchi, Comi, Connors, Milstein and Varsou2005; Chez et al., Reference Chez, Dowling, Patel, Khanna and Kominsky2007). Neuroinflammation, in general, is characterized by the reactivity of microglial cells and astrocytes, activation of inducible nitric oxide (NO)-synthase (i-NOS) and increased expression and/or release of cytokines and chemokines (Monnet-Tschudi et al., Reference Monnet-Tschudi, Defaux, Braissant, Cagnon and Zurich2011) and that is what is found in autism. As Herbert (Reference Herbert2005) pointed out in her review of the brain abnormalities in ASD, the autistic brain is not simply wired differently, but neuroinflammation is a part of the pathology in autism from childhood through adulthood.
For example, three post-mortem studies have shown microglial activation in ASD. First, Vargas et al. (Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005) examined brain tissue and cerebral spinal fluid (CSF) in those with autism. For the morphological studies, brain tissues from the cerebellum, midfrontal and cingulate gyrus were obtained at autopsy from 11 patients with autism. Fresh-frozen tissues from seven patients and CSF from six living patients with autism were used for cytokine protein profiling. The authors found active neuroinflammatory process in the cerebral cortex, white matter, and notably in cerebellum of patients with autism, with marked activation of microglia and astroglia. The authors stated that the CSF showed a unique proinflammatory profile of cytokines. The authors also stated that the pattern of cellular and protein findings suggests that the brain's own immune system (not immune abnormalities from outside the brain) and the neuroinflammatory process appears to be an ongoing and chronic mechanism of central nervous system (CNS) dysfunction.
Second, Morgan et al. (Reference Morgan, Chana, Pardo, Achim, Semendeferi and Buckwalter2010) examined the dorsolateral prefrontal cortex of male cases with autism (n = 13) and control cases (n = 9) and found microglial activation and increased microglial density in the dorsolateral prefrontal cortex in those with autism. They also noted process retraction and thickening, and extension of filopodia (small protrusions sent out from a migrating cell in the direction that it wants to move) from the processes. The authors stated that the microglia were markedly activated in 5 of 13 cases with autism, including 2 of 3 under age 6, and marginally activated in an additional 4 of 13 cases. The authors stated that because of its early presence, microglial activation may play a central role in the brain pathogenesis of autism.
Third, Tetreault et al. (Reference Tetreault, Hakeem, Jiang, Williams, Allman and Wold2012) immunocytochemically identified microglia in fronto-insular and visual cortex in autopsy brains of well-phenotyped subjects with autism and matched controls and stereologically quantified the microglial densities. They found that in the fronto-insular and visual cortex, individuals with autism had significantly more microglia compared to controls. The authors concluded that because they observed increased densities of microglia in two functionally and anatomically disparate cortical areas, microglia are probably denser throughout cerebral cortex in brains of people with autism.
EVIDENCE OF ASTROCYTIC ACTIVATION IN THE BRAIN IN AUTISM
Numerous studies have shown that glial fibrillary acidic protein (GFAP) levels are increased in autism. An autopsy report by Bailey et al. (Reference Bailey, Luthert, Dean, Harding, Janota and Montgomery1998), for example, found that the Purkinje cell loss in ASD was sometimes accompanied by gliosis and an increase in GFAP.
A study by Ahlsen et al. (Reference Ahlsen, Rosengren, Belfrage, Palm, Haglid and Hamberger1993) examined the levels of GFAP in the CSF of children with autism and found that their average GFAP level was three times higher than it was in the control group. The authors stated that the results could implicate gliosis and unspecified brain damage in children with autism. Laurence and Fatemi (Reference Laurence and Fatemi2005) examined levels of GFAP in the frontal, parietal and cerebellar cortices using age-matched autistic and control post-mortem specimens. GFAP was significantly elevated in all three brain areas. The authors stated that the elevated GFAP confirms microglial and astroglial activation in autism and indicates gliosis, reactive injury, and perturbed neuronal migration processes.
Rosengren et al. (Reference Rosengren, Ahlsén, Belfrage, Gillberg, Haglid and Hamberger1992) also found GFAP levels in CSF in children with autism were higher than those in normal control children of the same age range. The authors stated that the high levels of GFAP in combination with normal S-100 protein concentrations in CSF indicate reactive astrogliosis in the CNS.
Also, Fatemi et al. (Reference Fatemi, Folsom, Reutiman and Lee2008) investigated whether two astrocytic markers, aquaporin 4 and connexin 43, are altered in Brodmann's Area 40 (BA40, parietal cortex), BA9 (superior frontal cortex) and the cerebella of brains of subjects with autism and matched controls. The authors reported that the findings demonstrated significant changes in the two astrocytic markers in the brain from individuals with autism.
RESULTS OF EXTENDED MICROGLIAL AND ASTROCYTIC ACTIVATION
When microglia remain activated for an extended period, the production of mediators is sustained longer than usual. This increase in mediators contributes to loss of synaptic connections and neuronal death (Wood, Reference Wood and Wood2003). Streit et al. (Reference Streit, Mrak and Griffin2004) state that in the case of chronic neuroinflammation, the cumulative ill effects of microglial and astrocytic activation can contribute to and expand the initial neurodestruction, thus maintaining and worsening the disease process through their actions. Neuroinflammation generally refers to more chronic, sustained injury when the responses of microglial cells expand the neurodestructive effects (Streita, Reference Streita2006). Evidence suggests that the collateral neural damage can involve loss of connections in the brain (Gehrmann et al., Reference Gehrmann, Matsumoto and Kreutzberg1995). Underconnectivity is found in autism and will be discussed in the next section.
EVIDENCE OF ABNORMAL BRAIN CONNECTIVITY IN ASD
It is apparent from many studies that ASD involves the loss of critically important neuronal connections and networks (Just et al., Reference Just, Cherkassky, Keller, Kana and Minshew2007; Kana et al., Reference Kana, Keller and Cherkassky2009; Minshew and Keller, Reference Minshew and Keller2010; Di Martino et al., Reference Di Martino, Kelly, Grzadzinski, Zuo, Mennes and Mairena2011; Wass, Reference Wass2011). In a recent review of connectivity in ASD, Wass (Reference Wass2011) stated that there is ‘considerable convergent evidence suggesting that connectivity is disrupted in ASD’. From his review of the literature, he states that the evidence indicates long-distance under-connectivity, and that disruptions appear more severe in the later-developing cortical regions.
As a result, the functional connectivity among regions of autistic brains is diminished (Herbert et al., Reference Herbert, Ziegler, Makris, Filipek, Kemper and Normandin2004, Reference Herbert, Ziegler, Deutsch, O'Brien, Kennedy and Filipek2005; Herbert, Reference Herbert2005). For example, Damarla et al. (Reference Damarla, Keller, Kana, Cherkassky, Williams and Minshew2010) investigated the cortical underconnectivity theory in autism by examining the neural bases of the visuospatial processing in high-functioning autism. Using a combination of behavioral, functional magnetic resonance imaging (fMRI), functional connectivity and corpus callosum morphometric methodological tools, they found that the autism group had lower functional connectivity between the higher-order working memory/executive areas and the visuospatial regions (between frontal and parietal–occipital).
Ebisch et al. (Reference Ebisch, Gallese, Willems, Mantini, Groen and Romani2011), using fMRI, found reduced functional connectivity in ASD, compared with controls, between anterior and posterior insula and specific brain regions involved in emotional and sensory processing. Di Martino et al. (Reference Di Martino, Kelly, Grzadzinski, Zuo, Mennes and Mairena2011) found that children with ASD have abnormal functional connectivity between nearly all striatal subregions and heteromodal associative and limbic cortex previously implicated in the physiopathology of ASD (e.g. insular and right superior temporal gyrus).
Shukla et al. (Reference Shukla, Keehn, Lincoln and Müller2010) found fiber tract abnormalities in the corpus callosum (indicating impaired interhemispheric transfer), internal capsule and middle cerebellar peduncle and all three segments of the internal capsule in ASD. Boger-Megiddo et al. (Reference Boger-Megiddo, Shaw, Friedman, Sparks, Artru and Giedd2006) assessed midsagittal corpus callosum cross-sectional areas in 3–4 year olds with ASD compared to typically developing (TD) and developmentally delayed (DD) children. Although there was no difference in absolute size compared to TD, ASD callosums were disproportionately small when adjusted for increased ASD cerebral volume. The ASD clinical subgroup analysis revealed greater proportional callosum reduction in the more severely affected autistic disorder than in pervasive developmental disorder-not otherwise specified children. Just et al. (Reference Just, Cherkassky, Keller, Kana and Minshew2007) found that relevant parts of the corpus callosum, through which many of the bilaterally activated cortical areas communicate, were smaller in cross-sectional area in the autistic participants and that the size of the genu of the corpus callosum was correlated with frontal–parietal functional connectivity.
Particularly implicated in connectivity is the cerebellum, one of the most common sites of anatomic abnormality in autism (Courchesne, Reference Courchesne1997; Courchesne and Pierce, Reference Courchesne, Pierce and Ramachandran2002; Belmonte et al., Reference Belmonte, Allen, Beckel-Mitchener, Boulanger, Carper and Webb2004). The Purkinje cell is the main output cell in the cerebellum and it is significantly diminished in number in ASD (Palmen et al., Reference Palmen, van Engeland, Hof and Schmitz2004).
It is important to note that the connectivity issues are related to the symptomatology in ASD. Using electroencephalography (EEG) to assess dynamic brain connectivity, Barttfeld et al. (Reference Barttfeld, Wicker, Cukier, Navarta, Lew and Sigman2011), for example, found that the greater the abnormalities found in the connectivity in ASD, the worse the child's symptoms.
EVIDENCE OF INCREASED PROINFLAMMATORY CYTOKINE LEVELS IN THE BRAIN AND CSF IN AUTISM (TNF-α, IFN-γ, IL-1β, IL-8)
As mentioned earlier, there is evidence of proinflammatory cytokine profiles in the brain and cerebrospinal fluid of children with ASD (Vargas et al., Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005; Zimmerman et al., Reference Zimmerman, Jyonouchi, Comi, Connors, Milstein and Varsou2005; Chez et al., Reference Chez, Dowling, Patel, Khanna and Kominsky2007). Some specific examples are as follows. Li et al. (Reference Li, Chauhan, Sheikh, Patil, Chauhan and Li2009) showed that proinflammatory cytokines (tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF)), Th1 cytokine (IFN-γ) and chemokine (IL-8) were significantly increased in the brains of ASD patients compared with the controls. A study by Vargas et al. (Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005) demonstrated tumor growth factor-β1, derived from neuroglia, was significantly increased in the middle frontal gurus of autistic patients, while macrophage chemoattractant protein (MCP)-1, IL-6 and IL-10 were increased in the anterior cingulated gurus. In addition, using protein array approach, Vargas and colleagues also found that MCP-1, IL-6, IL-8, and IFN-γ were significantly increased in the CSF (Vargas et al., Reference Vargas, Nascimbene, Krishnan, Zimmerman and Pardo2005). TNF-α was also shown to be increased in the CSF of autistic patients by Chez et al. (Reference Chez, Dowling, Patel, Khanna and Kominsky2007). Chez et al. stated that the elevation of cerebrospinal fluid levels of TNF-α was significantly higher (mean = 104.10 pg ml−1) than concurrent serum levels (mean = 2.78 pg ml−1) in all the patients studied. They stated that the ratio was significantly higher than the elevations reported for other pathological states for which cerebrospinal fluid and serum TNF-α levels have been simultaneously measured and that this finding may provide an insight into CNS inflammatory mechanisms in autism.
POSSIBLE ROLE OF NF-κB EXPRESSION IN NEUROINFLAMMATION IN AUTISM
The neuroinflammation in autism appears to be chronic and even excessive. Recent research suggests that the exaggerated brain immune response in ASD may be due, in part, to aberrant nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB or NF-kappaB) expression, which can produce chronic or excessive inflammation (Young et al., Reference Young, Campbell, Lynch, Suckling and Powis2011). A recent study by Young et al. (Reference Young, Campbell, Lynch, Suckling and Powis2011) examined NF-κB in human post-mortem samples of orbitofrontal cortex tissue in autism as compared to controls. They hypothesized that the concentrations of NF-κB would be elevated, especially in activated microglia in ASD, and pH would be concomitantly reduced (i.e. acidification). According to the authors, neurons, astrocytes, and microglia all demonstrated increased extranuclear and nuclear translocated NF-κB p65 expression in brain tissue from ASD donors relative to samples from matched controls. The between-group differences were increased in astrocytes and microglia relative to neurons, but particularly pronounced for highly ‘mature microglia’. Measurement of pH in homogenized samples demonstrated a 0.98-unit difference in means and a strong (F = 98.3; P = 0.00018) linear relationship to the expression of nuclear translocated NF-κB in mature microglia. Young et al. (Reference Young, Campbell, Lynch, Suckling and Powis2011) summarized that NF-κB is aberrantly expressed in orbitofrontal cortex in patients with ASD, as part of a putative molecular cascade leading to inflammation, especially of resident immune cells in brain regions associated with the behavioral and clinical symptoms of ASD. Their study provides further evidence of neuroinflammation that may be categorized as excessive in ASD.
Naik et al. (Reference Naik, Gangadharan, Abbagani, Nagalla, Dasari and Manna2011) examined NF-κB in peripheral blood samples of 67 children with autism and 29 control children using electrophoretic mobility shift assay. They stated that there was a significant increase in NF-κB DNA binding activity in peripheral blood samples of children with autism and when the fold increase of NF-κB in cases (n = 67) was compared with that of controls (n = 29), there was a significant difference (3.14 versus 1.40, respectively; P < 0.02). They concluded that autism may arise, at least in part, from an NF-κB pathway gone awry.
Evidence suggests that the equivalent of a vicious cycle can occur where microglia produce oxidative products and then increased intracellular reactive oxygen species (ROS), in turn, activates a redox-sensitive NF-κB to provoke excessive neuroinflammation. According to Nakanishi et al. (Reference Nakanishi, Hayashi and Wu2011), this can result in memory deficits and prolonged behavioral consequences.
POSSIBLE ROLE OF GLUTATHIONE DEPLETION IN MICROGLIAL ACTIVATION IN AUTISM
Recent research suggests that glutathione (GSH) depletion can play a role in microglia-mediated neurotoxicity (Lee et al., Reference Lee, Mills, Schwartz, Bell, Deeg and Ruthazer2010). Lee et al. (Reference Lee, Mills, Schwartz, Bell, Deeg and Ruthazer2010), for example, explored whether GSH depletion stimulated a neuroinflammatory response. They found that inhibition of GSH biosynthesis with d,l-buthionine-S,R-sulfoximine causes human microglia and human astrocytes to release TNF-α, IL-6 and nitrite ions. They concluded that as astrocytes are a main supplier of GSH to microglia and neurons in the brain, depletion of GSH during aging or neurodegeneration in neurological diseases may not only lead to activation of microglia and the astrocytes themselves, but may also render neurons sensitive to cell death.
In addition, Kigerl et al. (Reference Kigerl, Ankeny, Garg, Wei, Guan and Lai2011) found that ex vivo analyses showed that redox balance in microglia and macrophages is controlled by induction of glutamate/cystine antiporter (system x(c)(-)) and that high GSH:GSSG ratios predict the neurotoxic potential of these cells. [Reduced glutathione (GSH) is a major tissue antioxidant. The formation of a disulfide bond between two GSH molecules gives rise to oxidized glutathione (GSSG). Under conditions of oxidative stress, GSSG accumulates and the ratio of GSH to GSSG will decrease. Therefore, the GSH/GSSG ratio can be used as an indicator of oxidative stress in cells and tissues.]
More importantly, studies suggest that children with ASD have inadequate GSH production. First, studies indicate abnormalities in the transsulfuration pathway in ASD (the pathway is where GSH is made) (James et al., Reference James, Cutler, Melnyk, Jernigan, Janak and Gaylor2004, Reference James, Melnyk, Jernigan, Cleves, Halsted and Wong2006; Geier et al., Reference Geier, Kern, Garver, Adams, Audhya and Geier2009). And studies show low plasma GSH levels in ASD (James et al., Reference James, Cutler, Melnyk, Jernigan, Janak and Gaylor2004, Reference James, Melnyk, Jernigan, Cleves, Halsted and Wong2006; Geier et al., Reference Geier, Kern, Garver, Adams, Audhya and Geier2009) and reduced glutathione regenerating enzymes (Al-Yafee et al., Reference Al-Yafee, Al-Ayadhi, Haq and El-Ansary2011). In addition, James et al. (Reference James, Rose, Melnyk, Jernigan, Blossom and Pavliv2009) used lymphoblastoid cells (LCLs) derived from autistic children and unaffected controls to assess relative concentrations of reduced glutathione (GSH) and oxidized disulfide glutathione (GSSG) in cell extracts and isolated mitochondria as a measure of intracellular redox capacity. Their results indicated that the GSH/GSSG redox ratio decreased and the percentage of oxidized glutathione increased in both cytosol and mitochondria in the autism LCLs.
More importantly, Chauhan et al. (Reference Chauhan, Audhya and Chauhan2012) compared DNA oxidation and glutathione redox status in post-mortem brain samples from the cerebellum and frontal, temporal, parietal and occipital cortex from autistic subjects and age-matched normal subjects. The authors reported that levels of reduced glutathione GSH were significantly reduced and the levels of oxidized glutathione GSSG were significantly increased in the cerebellum and temporal cortex in the brain samples from the group with autism as compared to the corresponding levels in the control brain samples.
Thus, it is possible that inadequate availability of GSH in ASD may play a role in microglial activation. Furthermore, GSH stores could be depleted from oxidative stress caused by microglial activation and if an individual cannot readily regenerate GSH, low GSH availability may stimulate microglial activation, leading to a cascade of events that potentiates itself.
In addition, in its resting state, microglia have been shown to contain levels of glutathione significantly higher than in astrocytes or neurons (Chatterjee et al., Reference Chatterjee, Noack, Possel and Wolf2000; Hirrlinger et al., Reference Hirrlinger, Gutterer, Kussmaul, Hamprecht and Dringen2000). It appears that the production of NO following microglial activation causes a decline in cellular GSH levels leading to brain oxidative damage (Moss and Bates, Reference Moss and Bates2001). A study by Heales et al. (Reference Heales, Bolaños and Clark1996) examined brain glutathione and nitric oxide synthase activity (which generates NO). They found that loss of GSH was accompanied by a significant increase in brain nitric oxide synthase activity, by up to 55%. Depletion of GSH in cultured neurons, by approximately 90%, led to a significant 67% increase in nitric oxide synthase activity, as judged by nitrite formation, and cell death. It was concluded by Heales and colleagues that depletion of neuronal GSH results in increased nitric oxide synthase activity.
EVIDENCE OF INCREASED NITRIC OXIDE PRODUCTION AND RELATED MEDICAL SYMPTOMS IN ASD
As mentioned earlier, once activated, microglia release large amounts of NO and superoxide as a cytotoxic attack mechanism (Colton and Gilbert, Reference Colton and Gilbert1987). ROS and RNS derived from NO and superoxide may also cause local cellular damage by reacting with proteins, lipids and nucleic acids (Valko et al., Reference Valko, Leibfritz, Moncol, Cronin, Mazur and Telser2007). These chemicals can directly damage cells and lead to neuronal cell death. As a result, elevated NO levels can cause a wide array of medical problems, many of which are found in ASD. Although an ASD diagnosis is defined by three core features (impairment in communication and socialization, and behavioral issues), other features, more physical or systemic in nature, are associated with an ASD diagnosis.
For example, a recent analysis of the National Health Interview Survey, 2006–2010, that included 375 children with autism by Schieve et al. (Reference Schieve, Gonzalez, Boulet, Visser, Rice and Braun2011), found that children with autism were more likely to have headaches/migraines, respiratory and food allergies, physician visits and to be taking prescription medication than children without autism. Children with ASD are also, according to a study by Atladóttir et al. (Reference Atladóttir, Thorsen, Schendel, Østergaard, Lemcke and Parner2010), more likely to be hospitalized for an infectious disease. These medical diseases could possibly be a result of, or associated with, or exacerbated by elevated NO levels. Some examples are as follows:
As mentioned, children with ASD have a higher rate of infection (Atladóttir et al., Reference Atladóttir, Thorsen, Schendel, Østergaard, Lemcke and Parner2010). There is evidence that abundant NO at an inflammatory site may reduce and impair natural killer (NK) cell function (Takabayashi et al., Reference Takabayashi, Kawai, Iwata, Kanai, Denno and Kawada2000) which may provide an explanation for the frequent infections that a large subset of children with autism suffer from (Nicolson et al., Reference Nicolson, Gan, Nicolson and Haier2007). Studies have found low NK function in ASD (Enstrom et al., Reference Enstrom, Lit, Onore, Gregg, Hansen and Pessah2009). Vojdani et al. (Reference Vojdani, Mumper, Granpeesheh, Mielke, Traver and Bock2008), for example, found that at least 45% of children with autism suffer from low NK cell activity.
Seizures are common in autism, occurring in 20–30% of patients based on the majority of studies. Epileptiform EEG abnormalities are present in 10.3–72.4% of patients (Danielsson et al., Reference Danielsson, Gillberg, Billstedt, Gillberg and Olsson2005; Kagan-Kushnir et al., Reference Kagan-Kushnir, Roberts and Snead2005). Several studies have found that microglial activation can result in seizures (Radewicz et al., Reference Radewicz, Garey, Gentleman and Reynolds2000; Somera-Molina et al., Reference Somera-Molina, Robin, Somera, Anderson, Stine and Koh2007, Reference Somera-Molina, Nair, Van Eldik, Watterson and Wainwright2009). In a study by Kovács et al. (Reference Kovács, Rabanus, Otáhal, Patzak, Kardos and Albus2009) the researchers propose that NO-dependent enhancement of synaptic transmission is a key promoting factor for the initiation of seizures. In addition, NO might exert long-term effects in epilepsy. NO-dependent inhibition of mitochondrial electron transport chain activity (Brown, Reference Brown2001), disruption of the mitochondrial networks (Yuan et al., Reference Yuan, Gerencser, Liot, Lipton, Ellisman and Perkins2007), and blockade of mitochondrial trafficking (Rintoul et al., Reference Rintoul, Bennett, Papaconstandinou and Reynolds2006) might contribute to metabolic impairment as described for the epileptic hippocampus (Kunz et al., Reference Kunz, Kudin, Vielhaber, Blümcke, Zuschratter and Schramm2000; Kann et al., Reference Kann, Kovács, Njunting, Behrens, Otáhal and Lehmann2005).
In the National Health Interview Survey, 2006–2010, Schieve et al. (Reference Schieve, Gonzalez, Boulet, Visser, Rice and Braun2011) also reported a higher rate of asthma and bronchitis in children with intellectual disabilities (ID), including ASD. In a review by Ashutosh (Reference Ashutosh2000), it was reported that an increase in the exhaled NO has been shown to accompany eosinophilic inflammation and to correlate with other indices of inflammation in asthma. Exhaled NO increases during exacerbation and decreases with recovery in patients with asthma. Yates (Reference Yates2001) also reported that asthma is characterized by chronic airway inflammation and increased synthesis of NO and other highly reactive and toxic substances (ROS). Pro-inflammatory cytokines such as TNF-α and IL-1β are secreted in asthma and result in inflammatory cell recruitment, but also induce calcium- and calmodulin-iNOS and perpetuate the inflammatory response within the airways. NO is released by several pulmonary cells including epithelial cells, eosinophils and macrophages, and NO has been shown to be increased in conditions associated with airway inflammation, such as asthma and viral infections.
Research suggests relatively high rates of eczema and food allergies in ASD as compared to TD children (Schieve et al., Reference Schieve, Gonzalez, Boulet, Visser, Rice and Braun2011). It has been suggested that NO is an important player in eczema, food allergies, and intestinal inflammation. Eczema is characterized by inflammation of the skin and is commonly associated with food allergy. The results of a study, by Devenney et al. (Reference Devenney, Norrman, Forslund, Fälth-Magnusson and Sundqvist2010), were able to support previous studies indicating that the homeostasis of nitrogen radicals is disturbed in childhood eczema.
As mentioned, Adams et al. (Reference Adams, Johansen, Powell, Quig and Rubin2011) and Wang et al. (Reference Wang, Tancredi and Thomas2011) found that there is a correlation of gastrointestinal symptoms with autism severity indicating that children with more severe autism are likely to have more severe gastrointestinal symptoms and vice versa. Schieve et al. (Reference Schieve, Gonzalez, Boulet, Visser, Rice and Braun2011) found that children with autism were 70% more likely than children in the ID group, two times more likely than children in the attention-deficit hyperactivity disorder and learning disabled/other developmental delay groups, and seven times more likely than children without developmental delays (DDs) to have had frequent diarrhea/colitis in the last 12 months. Research shows that exaggerated or uncontrolled expression of iNOS itself becomes detrimental to the gastrointestinal tract (Calatayud et al., Reference Calatayud, Barrachina and Esplugues2001), and that large amounts of NO can increase gut permeability and induce apoptosis (Dijkstra et al., Reference Dijkstra, van Goor, Jansen and Moshage2004). Inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS) are chronic diseases that cause inflammation of the intestines. A study by Reinders et al. (Reference Reinders, Herulf, Ljung, Hollenberg, Weitzberg and Lundberg2005) found that NO was low in healthy control subjects, and variations over time were small. In IBS patients NO was slightly elevated, whereas patients with active IBD or collagenous colitis had greatly increased NO levels. Rectal NO correlated with disease activity in IBD and collagenous colitis and decreased markedly in IBD patients responding to anti-inflammatory treatment.
A statistically significant global reduction of cerebral blood flow (CBF) is found in autistic children (Burroni et al., Reference Burroni, Orsi, Monti, Hayek, Rocchi and Vattimo2008). Recent studies on brain circulation have provided evidence that CBF is impaired by decreased formation of NO from endothelial cells, autonomic nitrergic nerves or brain neurons and also by increased production of ROS. The NO–ROS interaction is an important topic in discussing blood flow and cell viability in the brain (Toda et al., Reference Toda, Ayajiki and Okamura2009).
In the recent study, Giulivi et al. (Reference Giulivi, Zhang, Omanska-Klusek, Ross-Inta, Wong and Hertz-Picciotto2010) found that children with autism were more likely to have mitochondrial dysfunction than TD children. Evidence has also been provided that mitochondrial dysfunction can be induced by elevated levels of NO (Stewart and Heales, Reference Stewart and Heales2003). NO and its toxic metabolite peroxynitrite (ONOO(-)) can inhibit the mitochondrial respiratory chain, leading to energy failure and ultimately cell death. ROS and RNS derived from NO and superoxide may also inhibit mitochondrial brain energy metabolism (Valko et al., Reference Valko, Leibfritz, Moncol, Cronin, Mazur and Telser2007), preventing the production of adenosine triphosphate (Bolaños et al., Reference Bolaños, Heales, Land and Clark1995).
Abnormal eating patterns and eating disorders are associated with ASD (Maenner et al., Reference Maenner, Arneson, Levy, Kirby, Nicholas and Durkin2011; Tang et al., Reference Tang, Piazza, Dolezal and Stein2011). A team of researchers in Italy provided evidence on the possible actions of NO on the etiology of eating disorders (Vannacci et al., Reference Vannacci, Ravaldi, Giannini, Rotella, Masini and Faravelli2006). In this study, plasma nitrite and cyclic guanosine monophosphate levels were significantly higher in eating disorder patients than in healthy controls.
CONCLUSION
Evidence indicates that children with ASD suffer from an ongoing neuroinflammatory process in different regions of the brain involving microglial activation. The microglial activation appears to play a role in the brain underconnectivity and other issues in ASD (see Fig. 1 which shows the relationships and interplay between microglial activation and the neuropathology, medical issues and symptoms in ASD). Current therapies typically used in ASD do not directly address the underlying neuroinflammation. It is possible that by reducing brain inflammation and microglial activation, the neurodestructive effects of chronic inflammation could be reduced and allow for neuronal reconnection. Reducing brain inflammation could allow for an improved response to early behavioral and learning intervention measures to be more effective, and ultimately enhance developmental outcomes. Many studies suggest that there are pharmaceutical and nutraceutical treatments that can reduce microglial activation and/or their associated inflammatory cytokines. However, the studies that have examined these potential therapies have not been conducted in ASD. Future studies that examine treatments that may reduce microglial activation and neuroinflammation, and ultimately help to mitigate symptoms in ASD, are warranted.
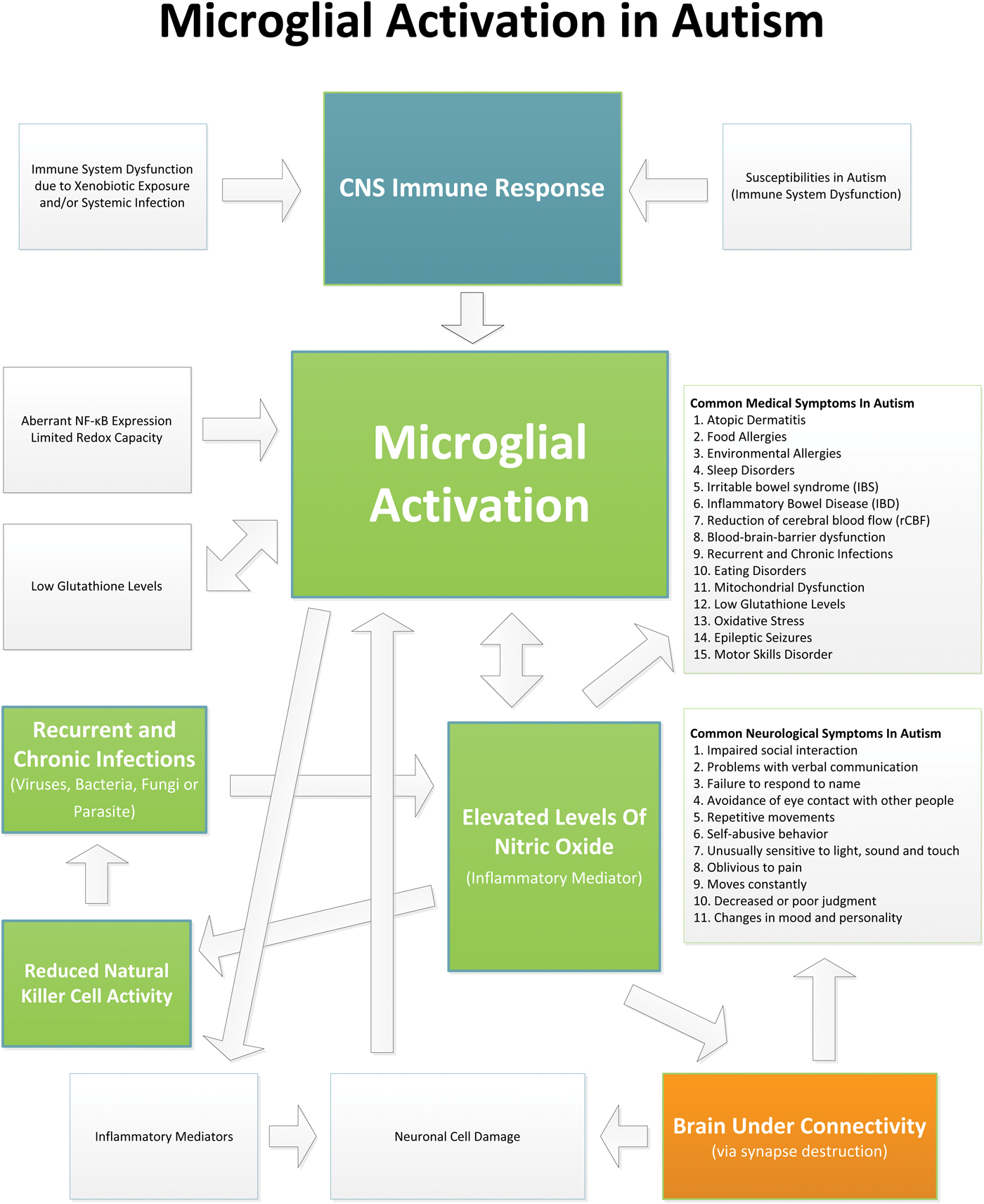
Fig. 1. This diagram shows the relationships and interplay between microglial activation and the neuropathology, medical issues and symptoms in ASD.
Statement of interest
None.


 Open access
Open access