


INTRODUCTION
Crimean Congo haemorrhagic fever virus (CCHFV) is a member of the family Bunyaviridae, genus Nairovirus [Reference Elliott, Regenmortel, Fauquet, Bishop, Carstens, Estes, Lemon, Maniloff, Mayo, McGeoch, Pringle and Wickner1]. This tick-borne zoonotic virus is associated with clinical disease ranging from a non-specific febrile illness to severe disease manifesting as haemorrhagic fever. CCHFV has a negative-sense, single-stranded RNA genome consisting of three segments designated large (L), medium (M) and small (S). The highly conserved complementary terminal nucleotide sequences result in loosely circular RNAs which together with the nucleocapsid protein, make up the three helical nucleocapsids. The L segment encodes the viral RNA polymerase while the S segment encodes the viral nucleocapsid protein. The M segment encodes a polyprotein which undergoes proteolytic processing to yield the viral glycoproteins, Gc and Gn [Reference Schmaljohn, Hooper, Knipe and Howley2].
CCHFV has been documented in more than 30 countries of Africa, Asia, Europe and the Middle East, with a distribution following that of Hyalomma ticks, the principal vector of the virus [Reference Whitehouse3, Reference Bente4]. Since the first description of clinical disease due to CCHFV infection in South Africa in 1981, 192 cases have been confirmed [Reference Msimang5]. However, complete S, M and L sequences are available for only four isolates collected from humans and one tick isolate from South Africa. These, as well as other published CCHFV sequences, were determined by Sanger sequencing using primer walking [Reference Aradaib6–Reference Zhou18]. In recent years, a number of next-generation sequencing (NGS) methods have been developed which yield large amounts of sequencing data at relatively low cost. Although the specifics differ, these methods all make use of three steps, namely, library preparation, DNA capture and enrichment, and sequencing or detection. In the field of virology, these techniques have been employed for various purposes including the discovery of novel viruses, whole viral genome sequencing, and ‘deep’ sequencing to determine viral quasi-species or genome variability [Reference Capobianchi, Giombini and Rozera19]. This study aimed to make use of NGS techniques to obtain whole genome sequences of southern African CCHFV isolates in order to perform genetic analysis including identification of reassortment events.
METHODS
Viral isolates
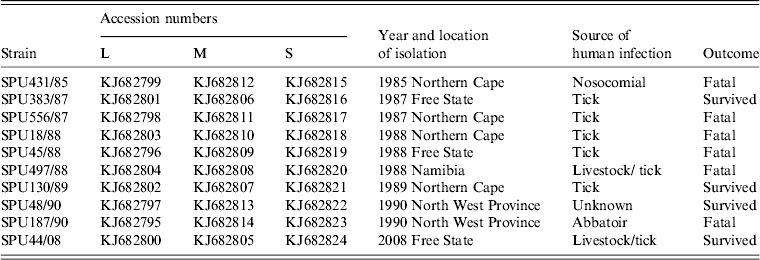
Ten CCHFV isolates obtained from patients in southern Africa between 1985 and 2008 were sequenced retrospectively. RNA was extracted and supplied by Professor J. T. Paweska, Centre for Emerging and Zoonotic Pathogens, National Institute for Communicable Diseases (NICD), Johannesburg from cell culture preparations or mouse brain suspensions. Total RNA was extracted for isolate SPU44/08 using TRIzol® reagent (Invitrogen, USA) according to the manufacturer's instructions. The remainder of the isolates were stored at −70°C as freeze-dried 10% suckling mouse brain suspensions at the level of mouse brain passage 2–3. The suspensions were inoculated into Vero cell cultures and total RNA extracted from the infected cells using the acid guanidium thiocyanate-phenol-chloroform method as described previously [Reference Burt and Swanepoel20]. The extracted RNA was stored at −70°C until use.
Reverse transcriptase–polymerase chain reaction (RT–PCR) and sequencing
The complete L segment was amplified using two previously described primer pairs that generated overlapping amplicons [Reference Deyde8]. The complete M segment was amplified using primers designed by Deyde et al. [Reference Deyde8]. Existing sequences in GenBank were used to design primers SF1 and SR3 (Table 1) for amplification of the S segment. The respective forward primers were used to perform the reverse transcription step for each amplicon using SuperScript™ III Reverse Transcriptase (Invitrogen). PCR was performed with the Expand Long Template PCR System (Roche Diagnostics GmbH, Germany) using standard cycling conditions according to the manufacturer's instructions and an annealing temperature of 48°C. The PCR products were gel extracted and purified using the Wizard® SV Gel and PCR Clean-Up System (Promega, USA).
Table 1. PCR and sequencing primers utilized for generation of amplicons used for sequencing

* Primers designed for sequence determination.
The complete genomes for 3/10 isolates (SPU431/85, SPU383/87, SPU 130/89) were determined using the Ion Torrent PGM™ sequencer (Life Technologies, USA) by the Central Analytical Facility, Stellenbosch University. For the remaining seven isolates, the S segment data was determined previously in the laboratory using the Big Dye™ Terminator v. 3.1 Cycle Sequencing kit (Applied Biosystems, USA) according to the manufacturer's instructions and three overlapping primer sets as described in Table 2. The L and M segments of these isolates were sequenced at the NICD, Johannesburg using the Roche 454 GS Junior™ sequencing system (Roche Diagnostics GmbH). Additional sequencing of all isolates for incomplete coverage and clarification of ambiguities, including mixed bases or nucleotide substitutions and base insertions or deletions, was performed as required using the Big Dye™ Terminator v. 3.1 Cycle Sequencing kit (Applied Biosystems). Details regarding the additional primers used are available from the corresponding author upon request.
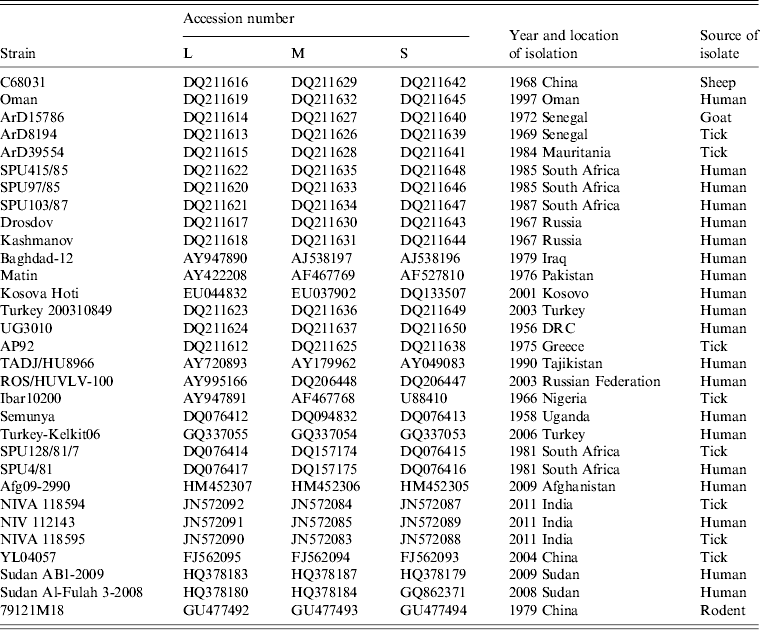
Table 2. Summary of data concerning the CCHFV sequences retrieved from the GenBank database and used in the study
Data analysis
Raw sequencing data was converted from SFF format to FASTQ files using the sff_extract script (available as part of seq_crumbs at http://bioinf.comav.upv.es/) and trimming and filtering of reads based on length and quality scores was then performed using PRINSEQ [Reference Schmieder and Edwards21]. Sequences for the L, M and S segments available in Genbank were used to compile databases and separate the reads into L, M and S segment-related data using filter_by_blast (http://bioinf.comav.upv.es/seq_crumbs/available_crumbs.html). De novo assembly of the blast-filtered and unfiltered reads was performed using MIRA [Reference Chevreux, Wetter and Suhai22]. The resulting contigs from both methods were assembled in Geneious (Geneious v. 4.8.5, Biomatters, http://www.geneious.com/) and compared to known complete CCHFV sequences to identify areas of incomplete coverage or ambiguities which required further investigation. Ambiguities and homopolymers were investigated using Gap5 [Reference Bonfield, Smith and Staden23] and the majority could be clarified by evaluation of the quality scores at each relevant position. Where necessary, additional Sanger sequencing was performed and sequences were incorporated into the assembly using Geneious and ChromasPro v. 1.42 (Technelysium Pty Ltd, Australia). Alignments were confirmed by visual inspection with reference sequences and comparison with previously published partial sequences.
Complete genome sequence data for 31 isolates were retrieved from GenBank as summarized in Table 2. The sequences were aligned using Clustal X version 2.0 [Reference Larkin24] and analysed using Molecular Evolutionary Genetics Analysis (MEGA) version 5 [Reference Tamura25] with the bootstrap maximum-likelihood method with 1000 replicates. Sequence divergence was also determined using MEGA by calculating the average P distances within and between sequence groups as well as pairwise distances for nucleotide and deduced amino acid sequences.
RESULTS
Sequencing data
Full coverage of complete L, M and S segments was obtained for 10 southern African CCHFV isolates. The details of the 10 isolates are summarized in Table 3. Ion Torrent PGM sequencing yielded raw data of >80 000 reads including >13 million bases for each of the three isolates. Following stringent filtering and trimming of reads based on length as well as quality scores, >35 000 reads amounting to ∼7 million bases were included in the final analysis of each of these isolates. The 454 GS Junior sequencing data were more variable and duplicate runs were performed for four of the isolates in order to obtain complete coverage. The raw data for these seven isolates yielded between 2404 and 17 315 reads including 1–8 million bases per isolate. Following filtering and trimming, the final alignments were performed on between 1105 and 7042 reads with ∼500 000 to 3·5 million bases per isolate. The alignments generated by MIRA de novo assembly of both blast-filtered and unfiltered data corresponded, although improved contiguous quality was achieved with some isolates following the application of filter_by_blast. A comprehensive sequence database and optimization of blast parameters were required to ensure effective filtering of reads without loss of coverage, particularly of the highly variable M segment.
Table 3. Summary of southern African CCHFV isolates included in the study
Genetic analysis
The genetic relationship of isolates as determined by the maximum-likelihood method is shown for the S (Fig. 1 a), M (Fig. 1 b) and L (Fig. 1 c) segments. The phylogenetic groups were designated I–VII as defined previously [Reference Deyde8, Reference Carroll26]. The complete sequences confirm the phylogenetic grouping of southern African isolates SPU497/88, SPU130/89, SPU383/87, SPU18/88 and SPU45/88 and the occurrence of reassortment of the M segment of these isolates as suggested by partial sequencing [Reference Burt27]. The complete sequences also confirm that the incongruencies in M segment grouping were due to reassortment rather than recombination events. This pattern of M segment reassortment, showing clustering of the S and L segments with group III strains from South and West Africa while the M segment clusters with group IV isolates from Asia and the Middle East, was also noted for newly sequenced isolates SPU556/87, SPU44/08 and SPU431/85. Isolates SPU48/90 and SPU187/90 showed no evidence of reassortment, with all three segments falling within the group III South/West Africa lineage. Further evidence of M segment reassortment was seen in southern African isolates SPU97/85 and SPU415/85, as described previously [Reference Deyde8]. No evidence of S segment reassortment was seen in the available complete sequences, but L segment reassortment was seen in two CCHFV isolates from Senegal, ArD15786 and ArD8194 [Reference Deyde8]. The Chinese isolates YL04057 and 79121M18 appear to represent a new group as suggested by Zhou et al. [Reference Zhou18], with S segments related to group IV Asia and Middle East, while the M and L segments cluster separately from other known groups. Isolate SPU431/85 was obtained from the same patient but from a sample collected 3 days subsequent to previously published SPU415/85. The sequences correlated well with only a single non-synonymous mutation in the coding region of the L segment and four non-synonymous mutations in the coding region of the M segment, as confirmed by Sanger sequencing. RNA from both of these isolates was obtained from cell cultures and these additional passages may have contributed to the accumulated mutations noted.
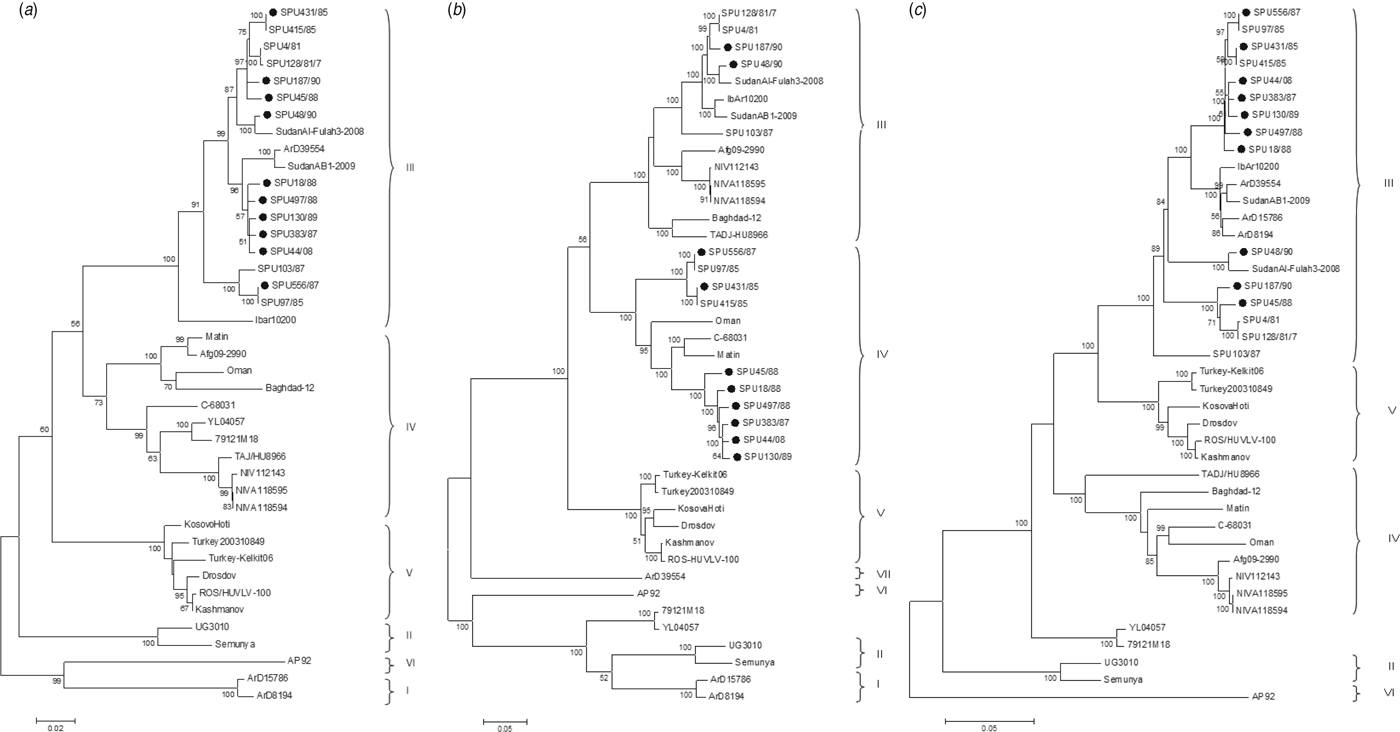
Fig. 1. Phylogenetic analysis of complete coding regions of (a) S segments, (b) M segments, and (c) L segments of CCHFV using a bootstrap maximum-likelihood method with 1000 replicates, with bootstrap values >50% indicated at the relevant nodes. Each sequence is designated by the isolate name and isolates sequenced in the current study are indicated by a solid circle (●).
The geographical distribution of CCHFV groups is illustrated in Figure 2. The S and L segments show a strong correlation in the distinct geographical grouping. The map displaying M segment group distribution clearly illustrates the blending of Asian and African strains, while the remaining groups correlate with S and L segment distributions.

Fig. 2. Geographical distribution of CCHFV groups for the (a) S segment, (b) M segment, and (c) L segment.
The mean P distances within groups and between groups as calculated with MEGA support the phylogenetic groupings. The S segment nucleotide distances within groups were generally low at between 0·1% and 1·4% and similar to the amino acid distances within groups which ranged from 0·1% to 1·8%. The nucleotide and amino acid distances between S segment groups were also similar at 2·5–6·8% and 2·8–8·4%, respectively. The L segment showed a similar degree of diversity within groups at both nucleotide and amino acid levels, but increased diversity between groups. Nucleotide distances within groups ranged from 0·4% to 2·5% and between groups from 3·8% to 11·3%, while amino acid distances within groups were between 0·5% and 1·8% and amino acid distances between groups were 2·9–10·1%. The M segment showed the greatest genetic diversity, particularly between groups, including numerous non-synonymous mutations. The nucleotide distances within groups ranged from 0·4% to 5·5%, while the nucleotide distances between groups were between 10·9% and 26·8%. At the amino acid level, the distances within groups were between 1·1% and 8·3% and the distances between groups were between 15·1% and 30·1%.
Among the southern African isolates, the S segment was highly conserved with both nucleotide and amino acid mean distances of <1% (range 0–1·3% and 0–1·7%, respectively). The diversity was also low for the L group with mean distances of 1·7% at the nucleotide level (range 0–2·8%) and 1·2% at the amino acid level (range 0–2·1%). The diversity of M segments was higher in group IV isolates than group III, with mean nucleotide distances of 5% and 2·8%, respectively and mean amino acid distances of 7·5% and 4·1%, respectively. Overall, the southern African M segments showed distances of 0–12·4% at the nucleotide level and 0–15·6% at the amino acid level.
Although no association between M segment reassortment and pathogenicity was noted in the southern African isolates included in the current study, the number of both non-fatal infections and non-reassortants included was small. Furthermore, no temporal association was seen with the isolation of reassortants.
DISCUSSION
The S segment of CCHFV was previously investigated as a surrogate for complete sequencing due to its length and the fact that it is relatively conserved in comparison to the M and L segments thereby simplifying the sequencing process. However, the demonstration of both reassortment [Reference Deyde8, Reference Burt and Swanepoel20, Reference Burt27, Reference Grard28] and, less commonly, recombination [Reference Lukashev29] among CCHFV genomes confirmed the necessity of at least partial sequencing of all three segments in order to perform accurate genetic analyses. The present study made use of two NGS platforms, namely the Ion Torrent PGM and the Roche 454 GS Junior sequencing systems, to obtain complete CCHFV sequences for ten southern African isolates. The largest collection of complete CCHFV genome sequences to date made use of 25 S, 40 M and 84 L primers in order to obtain complete sequence data for 13 geographically distinct isolates [Reference Deyde8]. NGS methods present a relatively simple alternative, requiring only appropriate primers for the generation of amplicons by RT–PCR and potentially a limited number of primers to verify isolated regions or bases. This process could be simplified further by performing NGS of CCHFV directly from clinical samples without prior amplification. This would not only negate the need for specific primers and therefore allow sequencing of diverse CCHFV isolates but would also remove bias introduced by PCR errors [Reference Bracho, Moya and Barrio30] as well as multiple passages sometimes required to generate adequate viral titres. This method has been successfully used to sequence RNA viruses such as lyssaviruses directly from tissue samples and cell culture lysates [Reference Marston31]. The introduction of errors during PCR can largely be overcome by using high fidelity enzymes. Although the assembly of raw data generated by NGS platforms can be complex, the workflow described in the present study made use almost exclusively of open source software and could be applied to a range of datasets from various sources. Confirmation of the validity of the assemblies obtained was made possible by comparison with a number of partial sequences which were available from a previous study [Reference Burt27], confirming the accuracy of the methods used.
Genetic evolution of arboviruses, including CCHFV, is a complex process influenced by multiple factors. As with other RNA viruses, the RNA-dependent RNA polymerases lack proofreading activity and show error frequencies of ∼10−4 [Reference Steinhauer, Domingo and Holland32]. This is offset by the effect of alternating infections of arthropods and vertebrates which constrains virus adaptation of arboviruses in comparison to other RNA viruses [Reference Coffey33]. Despite this, CCHFV shows a high level of genetic variability. Inclusion of diverse isolates in the current study, particularly from China and central and West Africa, led to nucleotide variation of up to 7%, 27% and 11% for the S, M and L segments, respectively, and amino acid variation of up to 8%, 30% and 10%. This is similar to previous studies with greater variability of the M and L segments compared to the S segment, which is contrary to expectation as the viral RNA polymerase is usually highly conserved [Reference Deyde8, Reference Anagnostou and Papa34].
Tree topologies for southern African isolates based on partial S, M and L sequence data correlated with topologies constructed using complete genome data (data available from corresponding author on request), provide further evidence that incongruencies in grouping are likely reassortant events and not recombination [Reference Burt27]. Genetic relationships indicate movement of CCHFV isolates within and between continents. Isolates from the same group can be found in geographically distinct locations and isolates from different groups can be found to co-circulate in similar regions. Genetic diversity within regions supports movement of the virus by bird migration and livestock trade. Reassortment events have occurred between West African and southern African isolates and between southern African and Asian isolates. Interestingly, the reassortment events between West Africa and southern Africa involved the L segment, whereas reassortment events between southern Africa and Asia involved the M segment. This may be related to the tick species found in these geographical areas. The mechanism of reassortment is unclear; however, it is assumed to occur in the tick host where dual persistent infections are more likely. Few complete sequences are available from West Africa but as three of the four available isolates show either L or M segment reassortment, it appears that reassortant viruses may occur frequently in this area. The two novel Chinese isolates described by Zhou et al. cluster independently in an as yet unnamed group, suggesting the occurrence of further genetic groups [Reference Zhou18]. Additional complete genome sequence data from geographically distinct isolates are required to corroborate and expand these findings.
Reassortment is widely described for members of the Bunyaviridae family [Reference Briese, Calisher and Higgs35]. Both homologous and heterologous reassortment may occur and may result in altered viral phenotypes. Ngari virus is a reassortant of Bunyamwera and Batai viruses and is associated with haemorrhagic fever, in contrast to the mild disease associated with the parent viruses [Reference Bowen36]. Both increased neuroinvasiveness and enhanced transmission by insect vectors have been specifically linked to the M segment of La Crosse virus, another member of the Bunyaviridae family [Reference Beaty37, Reference Gonzalez-Scarano38]. Burt et al. suggested a possible association between M segment reassortment of CCHFV and pathogenicity although this finding was not statistically significant [Reference Burt27]. The current study could not confirm this association but given the small number of isolates included, further investigation is warranted. As 10 of the 15 complete sequences of CCHFV from southern Africa show M segment reassortment, it would be of interest to determine whether these M segments provide a competitive advantage such as the increased transmissibility demonstrated in La Crosse virus reassortants. The postulated co-evolution of S and L segments, possibly related to functional interdependency [Reference Chamberlain7], may account for the relative scarcity of reassortment of these segments.
It is likely that both genetic drift and shift of CCHFV genomes occur chiefly during infection of insect vectors rather than in mammalian hosts due to the longer period of infection and increased likelihood of super-infection with more than one strain [Reference Kinsella11, Reference Hoogstraal39]. In contrast to the short period of viraemia seen in humans [Reference Shepherd40], ticks remain infected over much longer periods with transstadial and, more rarely, transovarial transmission playing an important role in virus perpetuation and therefore also virus evolution [Reference Hoogstraal39, Reference Gonzalez41, Reference Gordon, Linthicum and Moulton42].
In conclusion, NGS methods provide a simple and effective alternative to Sanger sequencing for the determination of complete CCHFV genomes. The application of these methods to clinical samples without prior PCR amplification would provide further advantages and should be validated on CCHFV isolates. Our genetic analysis of complete genome sequences confirms the high frequency of M segment reassortment in southern African CCHFV isolates. Further studies are indicated to elucidate the possible consequences of reassortment particularly relating to viral pathogenicity and transmissibility.
DECLARATION OF INTEREST
None.
ACKNOWLEDGEMENTS
We thank Anelda van der Walt, Central Analytical Facility, Stellenbosch University for advice regarding NGS analysis. This project was funded by the National Health Laboratory Service Research Trust, the Polio Research Foundation, South Africa, and University of the Free State Cluster funding.





