


Introduction
Although Alzheimer's disease (AD) is mainly diagnosed in the elderly, its pathophysiological processes begin several years prior to the onset of symptoms (Ref. Reference Sperling1).
Clinical AD is preceded by a long asymptomatic period, which has been divided into three stages: (1) an initial preclinical stage, (2) a second mild, but progressive, cognitive impairment (MCI) and (3) the final stage of clinical dementia due to AD (Refs Reference Sperling1, Reference Albert2, Reference McKhann3). Recently, researchers have increasingly focused on the characterisation of stages of AD risk, as these provide a critical opportunity for potential intervention (Ref. Reference Sperling1).
With ageing, there is a natural decline in cognitive skills. Thus, it may be difficult to discriminate between early cognitive changes due to AD and normal ageing process (Ref. Reference Caselli4). In that context, the first evidence of dementia may be the subjective cognitive decline (SCD), defined as a self-reported memory impairment with normal cognitive performance (Ref. Reference Jessen5). Complainers present a higher rate of conversion of SCD to either MCI or dementia (Ref. Reference Reisberg6). Thus, epidemiological studies pointed SCD as a predictor of cognitive decline (Refs Reference Amieva7, Reference Jonker, Geerlings and Schmand8) and as an independent risk factor for dementia (Ref. Reference Jessen9). In succession, the prodromal stage of dementia, MCI, has been defined as memory impairment beyond that expected for normal ageing (Ref. Reference Petersen10). Several MCI phenotypes have been associated with AD progression (Ref. Reference Espinosa11); however, amnestic MCI (aMCI) confers a higher risk of conversion (Ref. Reference Espinosa11).
The identification of MCI subjects, or even SCD who will convert to MCI or dementia, puts across an interesting strategy for secondary prevention of AD. In that sense, the biomarkers of β-amyloidosis and tau-mediated neuronal injury are detected in subjects with normal cognition (Ref. Reference Jack12). However, these biomarkers are not sufficient to produce the clinical symptoms of MCI and dementia or are not specific to AD either (Ref. Reference Sperling1). Furthermore, these biomarkers are not sensitive to disease progression (Ref. Reference Drago13). Hence, new approaches are required to improve the differentiation of SCD or MCI converters to AD.
AD's genetics has gained much attention since AD presents a heritability of up 70% (Ref. Reference Avramopoulos14). Recently, researchers have been striving towards the identification of new AD's genetic risk factors. In that sense, the identification of a genetic risk profile for pre-dementia stages may prove to be a powerful approach to select the candidate subjects to prevent or delay the disease progression during the early preclinical stages. If a fraction of SCD and MCI patients are in the pre-AD stages, the identification of an increased number of AD risk alleles as well as that of additional genetic factors specifically influencing SCD or MCI progression can be expected.
In this work, we reviewed the available information in the literature for genome-wide significant variants associated with AD and their involvement in preclinical, prodromal and dementia stages of AD. Additionally, we provide new meta-analysed data for apolipoprotein E (APOE) ε4 in preclinical and prodromal stages.
Methods: meta-analysis
Meta-analysis was performed for exploring the role of APOE ε4 in: (1) risk of MCI and (2) risk of SCD.
Dataset selection
Literature search was conducted in PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) using the following keywords: (1) for MCI: APOE, genetics, risk, mild cognitive impairment and excluding reviews; and (2) for SCD: APOE, SCD. A total of 301 articles were found for MCI and 32 for SCD.
We selected the studies meeting the following criteria: (1) case/control studies or longitudinal studies where it is possible to distinguish a sub-population of cases and a sub-population of controls; (2) studies that provide a complete definition of the participants; (3) studies that evaluated the APOE ε4 genotype as a risk factor leading to MCI or SCD, or provided the numbers of APOE ε4 genotypes or provided sufficient data to calculate them; and (4) studies that provided an odds ratio (OR) with 95% confidence interval (CI) as well as the P-value or provide sufficient data to calculate them. Finally, of the 301 articles found for MCI, 207 did not follow inclusion criteria, 29 showed sample overlapping and 41 had restricted access. A total of 24 articles and 23 668 individuals on MCI were finally included. In the case of SCD, 21 of the 32 articles did not follow the inclusion criteria and 3 showed sample overlapping; finally, a total of 8 articles and 6824 individuals were included in the meta-analysis.
Meta-analysis
Meta-analysis was conducted using the inverse variant method (fixed-effects model) in Ephisheet Excel application. In the case of heterogeneity, DerSimonian and Liard method (random-effects model) was used. Heterogeneity was considered significant when I 2 > 50% and P < 0.05. Meta-analysis results and forest plots were obtained using OpenMeta.
Dementia stage: genetic risk factors of AD
AD is a genetically heterogeneous disorder. From a genetic point of view, two patterns of inheritance have been linked to the genomic loci: the autosomal dominant and the polygenic. Traditionally, these patterns have been associated with early and late onset forms of the disease, respectively. However, based on the family history, AD can be subdivided into autosomal dominant, familial and sporadic (Ref. Reference Goldman15).
Autosomal dominant AD
The familial autosomal dominant pattern in AD represents ~1% of all the AD cases and is found almost exclusively in early onset AD (EOAD) (Ref. Reference Goldman15). It occurs in at least three individuals in two or more generations, with two of the individuals being first-degree relatives of the third (Ref. Reference Goldman15).
Linkage and candidate gene studies in EOAD families led to the identification of disease-causing mutations in β-amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes (Refs Reference Goate16, Reference Sherrington17, Reference Levy-Lahad, Wijsman and Nemens18). Most frequent mutations are shown in PSEN1 and APP loci, respectively, which present complete penetrance in contrast to PSEN2, which presents 95% penetrance (Ref. Reference Goldman15). These identifications promoted the formulation of amyloid cascade hypothesis, which is still considered as a possible disease mechanism. Despite that, there are EOAD families with negative screening for APP, PSEN1 and PSEN2 mutations supporting the existence of additional causal genes (Ref. Reference Raux19). In addition, it is seen that APOE ε4 genotype, the major genetic risk factor for Late-Onset Alzheimer's disease (LOAD) (Ref. Reference Corder and Saunders20), also modifies the risk of EOAD (Ref. Reference Genin21).
Presently, 262 pathogenic mutations have been identified: 42 in APP, 207 in PSEN1 and 13 in PSEN2 (http://www.molgen.ua.ac.be/ADMutations); no other genes have been associated with an autosomal dominant form of AD.
Familial AD (FAD)
FAD occurs in more than one individual and, at least, two of the affected individuals are third-degree relatives or closer (Ref. Reference Goldman15). Most of the FAD cases are LOAD, but the presence of early onset FAD may be caused by hidden autosomal-dominant AD mutations (Ref. Reference Goldman15).
Sporadic AD (SAD)
SAD occurs in isolated cases in families or cases separated by more than three degrees of relationship. SAD represents 75% of all AD cases and typically presents a LOAD chart (Ref. Reference Goldman15).
The commonest AD phenotype, LOAD
The genetic and molecular basis for the commonest AD phenotype, i.e. LOAD, remains widely unknown. However, important progress on the isolation of the loci associated with AD has been achieved in the past few years because of the emergence of the genome-wide association (GWAS) and exome studies.
The ε4 allele of the APOE gene was the first genetic variant associated with LOAD (Ref. Reference Corder and Saunders20), and it remains as the major risk factor for the disease until now.
Behind the APOE discovery, the candidate gene approach led to the identification of two clusters of single nucleotide polymorphisms (SNPs) in SORL1 gene (Ref. Reference Rogaeva22). Recently, this association has been validated by International Genomics Alzheimer's Project (IGAP) (Ref. Reference Lambert23). However, candidate gene approach did not show more successful outcomes.
Most discoveries arrived when the GWAS strategy was applied to the large case-controlled datasets. In the GWAS era, common variants located at CLU, PICALM, CR1, BIN1, ABCA7, CD2AP, CD33, EPHA1 and MS4A6A-MS4A4E loci were associated with LOAD (Refs Reference Harold24, Reference Seshadri25, Reference Hollingworth26).
To validate the original GWAS findings, replication studies were performed with many independent datasets. Consequently, CR1, PICALM, CLU and BIN1 signals have been replicated in Caucasians (Refs Reference Jun27, Reference Carrasquillo28), Caribbean Hispanics (Ref. Reference Lee29) or Asian individuals (Refs Reference Yu30, Reference Jin31). In addition, the EPHA1 and CD33 genetic variants were replicated in Caucasian subjects (Ref. Reference Carrasquillo32), and ABCA7 in African Americans (Ref. Reference Reitz33).
A recent meta-analysis developed by IGAP in 74 046 individuals of European ancestry confirmed previously reported GWAS signals (ABCA7, BIN1, CLU, CR1, CD2AP, EPHA1, MS4A6A-MS4A4E and PICALM) (Ref. Reference Lambert23). Nevertheless, the CD33 locus, previously associated with LOAD, did not reach the genome-wide significance in the replication stages (Ref. Reference Lambert23). Moreover, the IGAP meta-analysis identified 10 novel genetic regions associated with LOAD: CASS4, SLC2A4A-RIN1, FERMT2, HLA-DRB5-HLA-DRB1, INPP5, MEF2C, PTK2B, CELF1, NME8 and ZCWPW1 and also confirmed a candidate gene, SORL1 (Ref. Reference Lambert23). Follow-up studies of the IGAP results revealed additional locus namely TRIP4 (Ref. Reference Ruiz34) and a novel AD locus within the microtubule-associated protein tau (MAPT) region at 17q21.31 (Ref. Reference Jun35). Finally, the gene-wide analyses of the IGAP dataset identified TP53INP1 and IGHV1-67 as the novel AD loci (Ref. Reference Escott-Price36), and an independent meta-analysis identified ATP5H/KCTD2 as the LOAD risk signal (Ref. Reference Boada37). Most loci reported after the initial IGAP report would require independent replications confirming its plausibility.
Despite that, the GWAS approach presented a disadvantage, i.e. its inability to detect rare variants, which might be a source of functional variants with larger effects on the LOAD risk (Ref. Reference Jonsson38). This lack was covered by the implementation of genome and exome sequencing technologies. Thus, in the recent years, rare variants with a significant effect on the risk for LOAD have been identified in APP, TREM2, PDL3 and UNC5C loci (Refs Reference Jonsson38, Reference Guerreiro39, Reference Cruchaga40, Reference Wetzel-Smith41). However, more efforts are needed to confirm the original signals. Several studies have confirmed the reported association of TREM2 with LOAD (Refs Reference Benitez42, Reference Ruiz43). In addition, the existence of TREM2 variants associated with the Naso–Hakola disease (Ref. Reference Paloneva44) and frontotemporal dementia (Ref. Reference Thelen45) supports its role in neurodegeneration. Alternatively, the PLD3 variants’ replication did not replicate the previous effect or overall burden analyses (Refs Reference Hooli46, Reference Heilmann47). Therefore, prudence is required to define the genuine signals associated with rare variants.
At present, 28 genetic regions have been associated with LOAD (Table 1), but many of them still require independent validation. These genes can be divided into four major functional clusters: (i) amyloid beta (Aβ) metabolism (APOE, CLU, ABCA7, CASS4, SORL1 and APP), (ii) Tau metabolism (BIN1, SLC2A4A-RIN1, CASS4, FERMT2 and 17q21.31 MAPT region), (iii) synaptic function (PICALM, CD2AP, EPHA1, SLC2A4A-RIN1, MEF2C and ZCWPW1) and (iv) immune response and inflammation (CLU, CR1, EPHA1, MS4A cluster, ABCA7, HLA-DRB5-HLA-DRB1, INPP5; MEF2C, TREM2 and IGHV1-67). Seven identified loci do not have a well-established pathway (PTK2B, CELF1, NME8, TRIP4, ATP5H/KCTD2, UNC5C and TP53INP1) (Fig. 1).
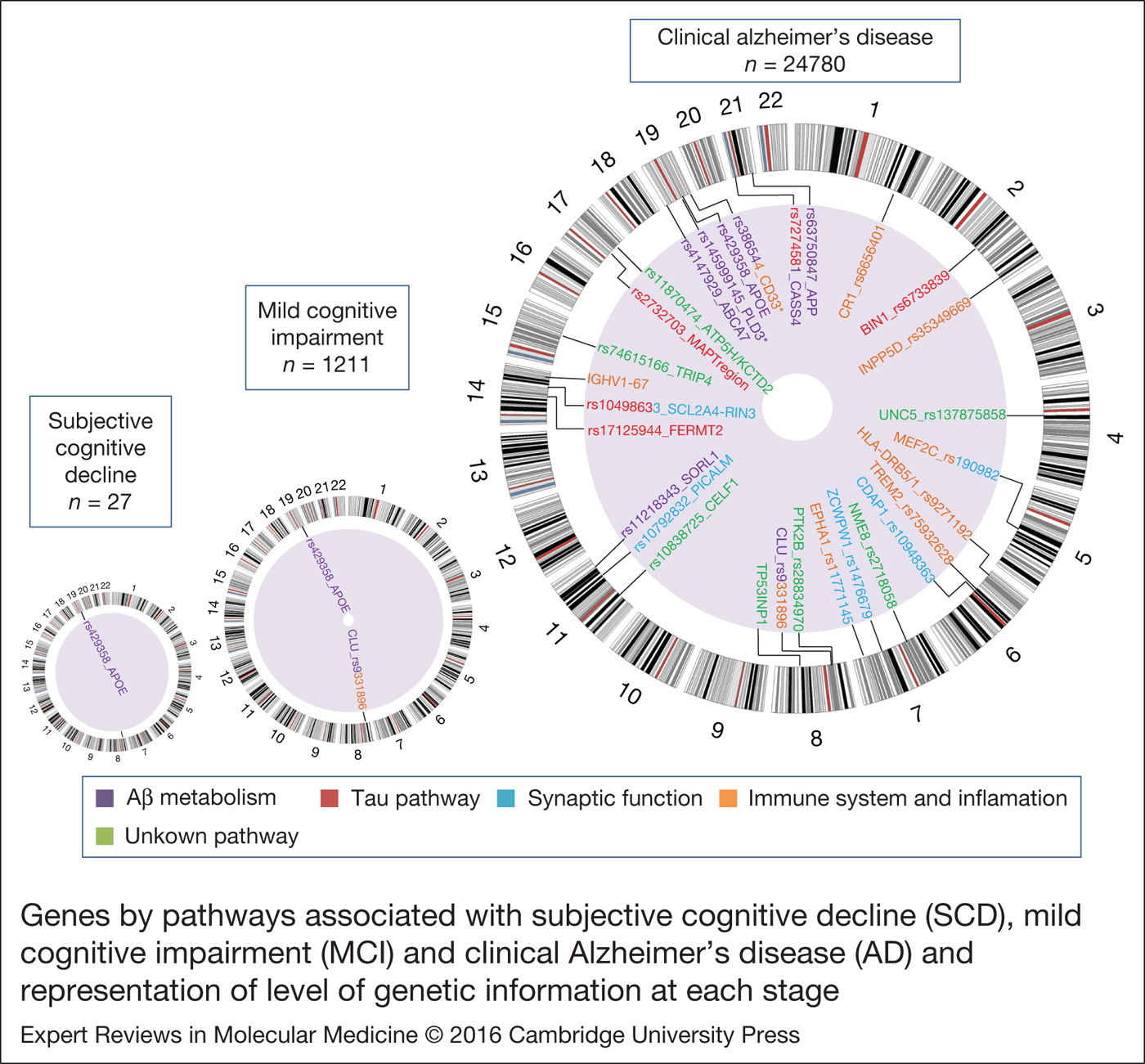
Figure 1. Genes by pathways associated with subjective cognitive decline (SCD), mild cognitive impairment (MCI) and clinical Alzheimer's disease (AD) and representation of level of genetic information at each stage. Genetic information response to the number of articles (n) found in the PubMed database with the keywords: Alzheimer's disease and genetics; mild cognitive impairment and genetics; subjective cognitive decline and genetics. Circular ideogram was performed using Circos (Ref. Reference Krzywinski120). *Not replicated in follow-up studies or in International Genomics Alzheimer's Project (IGAP).
Table 1. Genetic regions associated with LOAD from highest to lowest odds ratio.

a Not replicated in follow-up studies or IGAP. bEstimator extracted from Bertram et al. (Reference Bertram118). cEstimator extracted from Lambert et al. (Reference Lambert23).
IGAP: International Genomics Alzheimer's Project; LOAD: late onset Alzheimer's disease; NA: not available; OR: odds ratio.
Along with the identification of single locus, GWAS also permits the genetic confirmation of candidate pathways. Recently, pathway analysis studies have pointed toward the crucial role of the immune system in AD (Ref. Reference Lambert48), that has been further reinforced by the IGAP results (Ref. 49). Moreover, the IGAP study also implicates the regulation of endocytosis, cholesterol transport and protein ubiquitination as prime targets in the aetiology of AD (Ref. 49). The knowledge of the biological pathways involved in disease aetiology is crucial in the development of therapeutic strategies to aid in the prevention or treatment of LOAD.
Prodromal stage: genetics of mild cognitive impairment syndrome
APOE genotype in MCI
Petersen et al. (Ref. Reference Petersen50) were the first to provide evidence that MCI subjects with at least one allele of APOE ε4 presented a higher probability of conversion to dementia. Although subsequent genetic studies supported it (Refs Reference Fleisher51, Reference Artero52), they had small sample sizes, which only succeeded in providing an approximate value of the risk effect (Ref. Reference Fei and Jianhua53). Thus, the meta-analysis conducted by Elias-Sonnenschein et al. (Ref. Reference Elias-Sonnenschein54) provided the first consistent data corroborating the role of APOE ε4 as a genetic risk factor for progression from MCI to AD (Table 2).
Table 2. Genetic variants associated with risk or progression to dementia in MCI and SCD subjects.

a Amnestic MCI.
b Parameter of association measure hazard ratio; MCI: mild cognitive impairment; SCD: subjective cognitive decline; OR: odds ratio.
The involvement of APOE ε4 as a risk factor for MCI remains less explored. Therefore, here we have explored the risk conferred by APOE ε4 genotype to suffer MCI. Our meta-analysis, which includes 23 668 individuals of different ethnic groups, confirmed a significant risk association of APOE ε4 genotype and MCI [OR = 1.849 (1.587–2.153)] (Fig. 2) (Table 2). Our dataset showed high heterogeneity (I 2 = 63%; P-value < 0.001). In that sense, sub-population study revealed higher heterogeneity for Caucasian dataset with respect to Asiatic group (I 2 = 68%; P-value < 0.001; I 2 = 51%, P-value = 0.104). It must be considered that the major number of available studies is provided for Caucasians. Additionally, several studies have shown that the risk is higher for aMCI subpopulation in comparison with the rest of the MCI subtypes (Ref. Reference Lopez55) (Table 2). This makes sense in the context of MCI as the prodromal stage of AD.
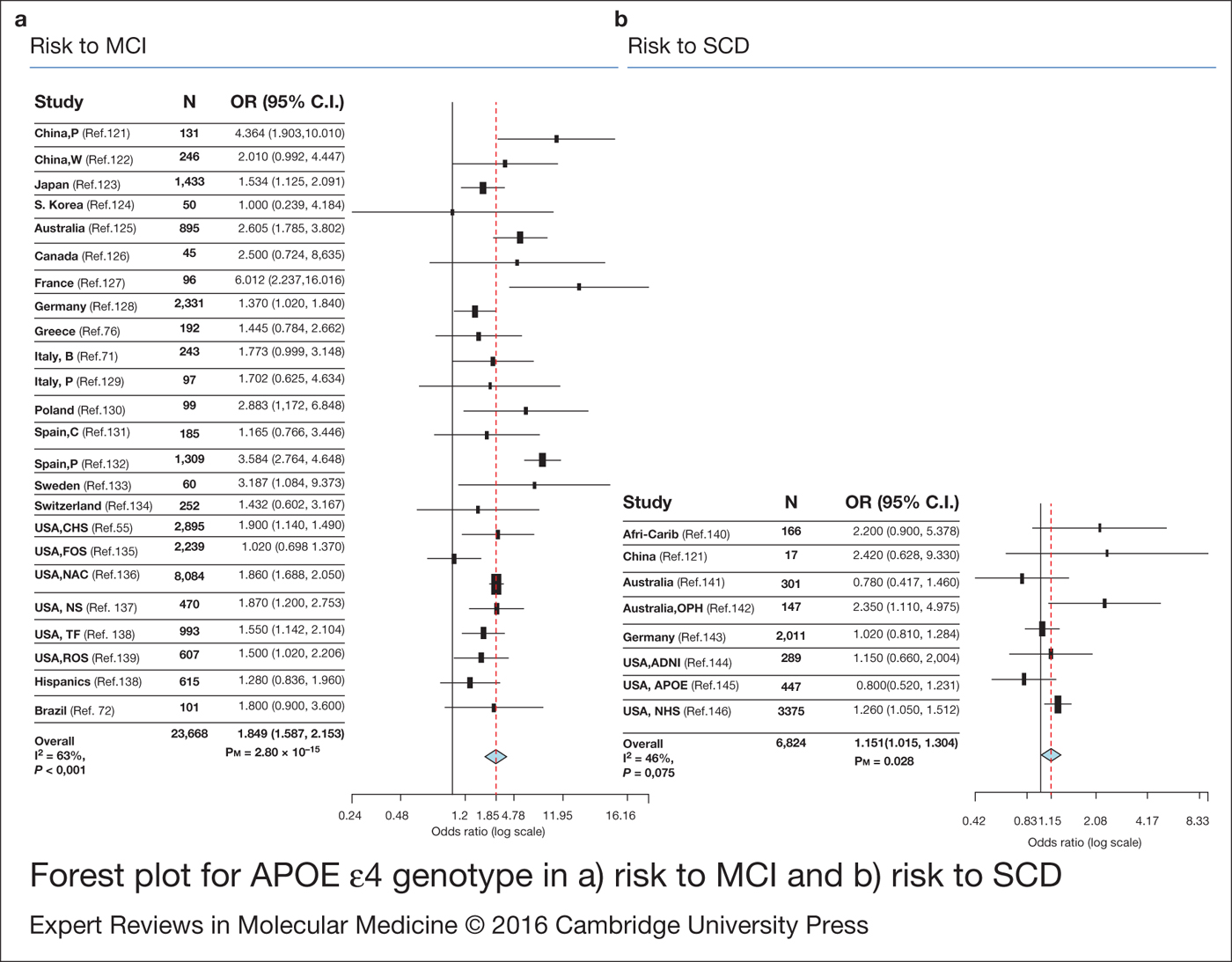
Figure 2. Forest plot for APOE ε4 genotype in (a) risk to mild cognitive impairment (MCI) and (b) risk to subjective cognitive decline (SCD). OR: odds ratio.
Although most of the attention has been focused on the risk allele ε4 of APOE, the ε2 allele has also demonstrated its role in AD (Ref. Reference Corder56). Some studies have shown that cognitively normal carriers of ε2 allele were less likely to present with cognitive decline (Refs Reference Wilson57, Reference Blacker58) and develop AD [relative risk (RR) = 0.76 (0.40–1.44)] (Ref. Reference Wilson57). Others showed that the MCI patients with ε2 allele had a better memory in comparison with non-carriers (Ref. Reference Bonner-Jackson, Okonkwo and Tremont59). Similarly, a recent study also detected the protective effect of APOE ε2 against pheno-conversion from MCI to AD [HR = 0.69 (0.51–0.86), P = 0.004] (Ref. Reference Lacour60). These observations suggest the role of ε2 allele in the protection against LOAD and its importance as a possible mechanism to reverse the APOE ε4 effect (Ref. Reference Lacour60).
Non-APOE LOAD loci in MCI
Apart from APOE, the genome-wide significant variants for LOAD in MCI population remain largely unknown. However, in the past few years, several studies have pointed out their influence in cognitive decline. Thus, CR1 and ABCA7 genes have been associated with faster rate of cognitive decline (CR1 P-value = 0.011; ABCA7 P-value = 0.013) (Refs Reference Chibnik61, Reference Carrasquillo62). EPHA1 and PICALM loci were also associated with faster and slower rate of decline (EPHA1 P = 0.013; PICALM P = 0.027), respectively, however they did not support Bonferroni corrections (Ref. Reference Hu63). Following studies for EPHA1 only replicated it marginally (P = 0.05) (Ref. Reference Carrasquillo62). Finally, the CLU locus was associated with cognitive endophenotypes in several studies. In that sense, the CLU risk allele has been associated with a faster rate of decline in some neuropsychological characteristics such as verbal immediate (P = 0.0032) and delayed free recall (P = 0.032) (Ref. Reference Thambisetty64) and its protective allele with a decreased risk of conversion to AD [OR = 0.25 (0.07–0.84), P = 0.025] (Ref. Reference Rodríguez-Rodríguez65). A study that evaluated the progression of normal subjects (n > 2000) to MCI/LOAD pointed the significant effect of CLU on logical memory delayed recognition (LMDR) (coefficient for LMDR = −0.51 (−0.92 to −0.11, P = 0.012) (Ref. Reference Carrasquillo62). It further showed a borderline significant hazard ratio (HR) in the sensitivity analysis between CLU risk allele and the risk of progression from MCI to AD (HR = 1.10, P = 0.13; sensitivity HZ = 1.14, P = 0.049) (Ref. Reference Carrasquillo62). Recently, another experiment conducted on 3326 MCI subjects of four countries supported the association of the CLU locus with the conversion from MCI to AD [HR = 1.19 (0.05–1.32), P = 0.0035] (Ref. Reference Lacour60) (Table 2).
Most of the identified LOAD loci present small risk effects and are, therefore, not quite informative for risk prediction on their own (Ref. Reference Rodríguez-Rodríguez65). Consequently, it is apprehended that the use of the genetic risk score (GRS) strategy, where multiple loci with modest effects are combined, might improve the identification of people at risk for common diseases (Ref. Reference Rodríguez-Rodríguez65). It was observed that MCI carriers of six or more non-APOE LOAD risk alleles showed rapid conversion to AD (Ref. Reference Rodríguez-Rodríguez65). However, in another study, the significant effect was only reached when the APOE genotype was considered (HR = 1.29, P = 1.14 × 10−9; sensitivity HR = 1.32, P = 5.73 × 10−10) (Ref. Reference Carrasquillo62). In a recent study, GRS for 19 LOAD loci with genome-wide significance was associated with MCI (OR = 1.15, P = 0.011) and with progression from MCI to dementia (HR = 1.59, P < 0.001) (Ref. Reference Adams66).
With the exception of the APOE and CLU variants, it seems difficult to arrive at a conclusion about the role of LOAD SNPs in the context of MCI. Studies developed in larger cohorts are needed to check and validate the expected association between the LOAD risk genetic variants and MCI.
New loci associated with MCI
It has been observed that additional loci have been associated with MCI. Recently, a GWAS associated rs12752888 (ACOT11 gene), rs7840202 (UBR5 – RRM2B region) and rs11637611 (unknown gene) markers with MCI progression (Ref. Reference Hu63) (Table 2). However, establishing a relationship with the present pathophysiological hypothesis of AD, at this stage, seems complex. Consequently, these signals require validation if they were to be discarded as false positive and either to be accepted as factors responsible for MCI progression.
Alternatively, candidate gene studies have suggested several aspirant genes associated with the pheno-conversion of MCI to AD. One such example is the MAPT gene. Specifically, a study revealed that the H1/H1 haplotype carriers presented a higher conversion rate of MCI to dementia (Ref. Reference Samaranch67). Moreover, the H1 haplotype has been associated with the risk of aMCI in converters to AD and non-converters (Ref. Reference Di Maria68) (Table 2). Recently, the region17q21.31, where MAPT was found, has been associated with LOAD reaching GWAS significance in non-APOE carriers (Ref. Reference Jun35). However, it remains unknown whether the casual variant is located in the MAPT or the nearby genes (Ref. Reference Jun35). For this reason, the MAPT linkage with MCI was explained at this point. Although MAPT is expected to be associated with LOAD because of the role of Tau protein in the classical AD hallmarks and the existence of several studies pointing towards this relationship (Refs Reference Cruchaga69, Reference Myers70), prudence is required until the location of the causal signal is identified.
Other genes associated with the pheno-conversion of MCI to AD are the vascular endothelial growth factor (Ref. Reference Chiappelli71), the brain-derived neurotrophic factor (BDNF) (Ref. Reference Forlenza72) or butyrylcholinesterase (BCHE) (Ref. Reference Ferris73). An additional marker in the serotonin transporter (5HTT) gene has also been related to MCI (Ref. Reference Marini74). It has also been associated with an emotion-induced retrograde amnesia (Ref. Reference Strange75), highlighting the role of serotonin in the memory system. The last reported association with MCI is detected in the α2b-adrenergic receptor (ADRAP2B) [OR = 0.491 (0.268–0.899); P = 0.021]; this association is also identified in AD subjects [OR = 0.463 (0.261–0.822); P = 0.009] (Ref. Reference Koutroumani76) (Table 2). Since none of these genes have been validated for LOAD, it can be said that they may act as genetic progression factors. They are capable of modulating the rate of decline but are not involved in the risk leading to AD.
Preclinical stage: SCD
There is limited research on the genetic variants that determine the risk to SCD or the progression of SCD to MCI or AD. Therefore, we have conducted the first meta-analysis exploring the involvement of APOE ε4 in the risk to suffer SCD. A significant risk effect was detected [OR = 1.151 (1.015–1.304)] (Fig. 2) (Table 2), with a borderline non-significant heterogeneity (I 2 = 46%, P-value = 0.075), which remains when the analysis is only performed for Caucasians (I 2 = 48%, P-value = 0.087). However, this significant association disappears [OR = 1.158 (0.933–1.437); P = 0.184] when a random model is used to conduct the meta-analysis. From our point of view, these results must be taken with prudence. SCD individuals represent a mixed population, where a pool of subjects may develop dementia, not exclusively AD and others never develop it. Hence, the sample size needed to detect Alzheimer's genuine genes must be larger.
Apart from the APOE ε4 polymorphism, other markers have been investigated to assess their possible association with SCD, such as alpha-2 macroglobulin gene (Ref. Reference Zill77), presenilin-1 mutation Glu318Gly (Ref. Reference Laws78), gene polymorphisms involved in vascular alterations (Ref. Reference Watfa79) and inflammatory genes (Ref. Reference Lau80). However, all these studies did not report any association with SCD.
The genetic profile of the SCD subjects is unexplored in spite of the fact that its analysis could provide new ways to manage the disease. The generation of large SCD datasets integrating genomic information with follow-up data would be an essential step in identifying genetic elements responsible for the progression of SCD to MCI and AD.
Other approaches: endophenotype-based approach
The use of quantitative traits closely related to the disease state, namely, endophenotypes, has been proposed as a simpler way to deal with genetic testing of LOAD. Thus, several endophenotypes have emerged across the cognitive spectrum of AD.
Differential amyloid burden and brain volume as endophenotype
Greater amyloid positron emission tomographic (PET) uptake is detected in AD, MCI and SCD cases as well as healthy controls who are carriers of the ε4 allele of APOE gene (Refs Reference Fleisher81, Reference Risacher82). APOE ε4 carriers become positive for amyloid PET imaging earlier (Ref. Reference Fleisher81) and show a higher cognitive decline (Ref. Reference Mormino83). Moreover, signals in APOE locus have been detected by GWAS of longitudinal studies for change in amyloid burden (Ref. Reference Ramanan84). Thus, in the past few years, APOE contribution to the determination of AD dementia converters has been reinforced. In addition, recent GWAS of longitudinal studies has also provided novel genetic correlations with the amyloid burden, such as BCHE, TREM1 and ILR1RAP (Refs Reference Mormino83, Reference Replogle85, Reference Ramanan86).
Findings around differential brain volumes have also involved LOAD loci showing that a reduced hippocampal volume (HCV) is associated with SORL1 in AD patients (Ref. Reference Louwersheimer87) and CLU gene in young healthy controls (Ref. Reference Lancaster88). HLA-DRB1 locus was correlated with a decrease in total brain volume along large longitudinal cohorts (Ref. Reference Chauhan89). Putamen volume was also associated with genetic variants, involved in apoptosis, axon guidance and vesicle transport (Ref. Reference Hibar90). In that context, axon guidance pathway was also associated with reduced HCV, as well as calcium and ErbB signalling (Ref. Reference Meda91). In addition, GRS for LOAD risk variants was associated with cortical thickness (Ref. Reference Sabuncu92) and reduced HCV in cognitively normal subjects, although HCV association disappears after removing APOE locus (Ref. Reference Chauhan89).
Amyloid-β and tau levels in cerebrospinal fluid (CSF)
CSF levels of Aβ42 and pTau181 have also been used as LOAD endophenotypes. APOE locus has been associated with both Aβ42 (Refs Reference Elias-Sonneschein93, Reference Cruchaga94, Reference Ramirez95) and pTau181 levels (Ref. Reference Cruchaga94). However, Elias-Sonnenschein et al. (Ref. Reference Elias-Sonneschein93) showed the correlations of APOE with Aβ42 but not with CSF tau biomarkers. Correlations with other LOAD loci remain scarce. Kauwe et al. (Ref. Reference Kauwe96) did not find any association for BIN1, CR1, CLU and PICALM, although, recently, CLU and MSA4A have been associated with Aβ42 levels (Ref. Reference Elias-Sonneschein93). In addition, GRS for AD loci did not provide any association (Ref. Reference Martinskainen97). Novel identifications have pointed to the 3q28 region, GLIS3 gene and TREM cluster association with tau biomarkers (Ref. Reference Cruchaga94) and SUCLG2 association with Aβ42 levels (Ref. Reference Ramirez95). For further information, refer to Cruchaga et al. (Ref. Reference Cruchaga, Ebbert and Kauwe98).
There is an inverse correlation between brain amyloid burden and Aβ CSF levels (Ref. Reference Fagan99). From our understanding, both techniques are dealing with the same pathological process, Aβ deregulation. In that scenario, the identification of the following two might be expected: (1) the same genetic factors independently of the analysed quantitative trait and (2) the reported LOAD loci associated with Aβ metabolism. Available data seem to be too far of these requisites, with the exception of APOE, the most consistent across studies and across AD stages. Therefore, from our view, an unsuited sample size affecting statistical power or the use of the incorrect endophenotypes of AD could be preventing new discoveries.
Brain genetic resistance factors: studies in healthy people
The presence of Alzheimer-type pathology in healthy elderly people at death (Ref. 100) evidenced the existence of compensatory mechanisms avoiding a cognitive decline in populations. A GWAS developed in this group of subjects suggested the involvement of the RELN in the compensatory mechanism for AD (Ref. Reference Kramer101) and illustrated that studies on non-demented subjects with AD neuropathology are an interesting starting point to identify brain genetic resistance factors.
The state of resistance to brain insults, where the neuropathological hallmarks without clinical AD existing, has been defined as the cognitive reserve (CR). Individuals with higher CR tolerate the pathology for a longer duration and show signs of cognitive decline later in life (Ref. Reference Stern102). Environmental and genetic factors are also believed to influence CR. It has been observed that the educational level (Ref. Reference Sando103), work complexity (Ref. Reference Andel104), engagement in leisure (Ref. Reference Scarmeas105), or social activity (Ref. Reference Wilson106) result in a reduced risk of dementia, contributing to CR. However, neuroplastic processes form the base of the above-mentioned factors. A study showed that the years of education is associated with genes involved in synaptic plasticity (Ref. Reference Rietveld107), and not surprisingly, cognition and neuroplasticity seem to be driven by shared genes (Ref. Reference Van Veluw108).
The human cognition has a heritable component (Refs Reference Deary, Johnson and Houlihan109, Reference Glahn110). Cognition status is associated with several genetic variants, such as genes involved in oxidative stress (Ref. Reference Harris111), biosynthesis of neurotransmitters (Ref. Reference Barral112), ubiquitin metabolism and immune system (Ref. Reference Debette113). It is to be noted that the immune system is highlighted as a prime pathway in LOAD (Ref. 49) and is expected to be linked with cognition. The association of memory with BDNF, 5HTT, and catechol-O-methyl transferase (COMT) genes remains more controversial (Ref. Reference Sabb114). However, as indicated, BDNF and 5HTT genes have also been associated with the conversion of MCI to AD (Refs Reference Forlenza72, Reference Marini74). These make them suitable candidates for further studies in genetics with MCI or SCD subjects.
Recently, two studies conducted on non-demented elderly subjects have showed that genes related to AD (TOMM40, APOE, MEF2C and ABCG1) are significantly associated with the cognitive function (Refs Reference Debette113, Reference Davies115). This suggests that genes involved in the normal and pathological cognitions somehow overlap (Ref. Reference Davies115) and highlight the applicability of the studies performed on healthy people. In addition, the cognition status has also been related to differential brain volumes (Ref. Reference Dowling116), thus, it seems that the HCV is a key component of the neuroanatomical basis of CR against memory in multiple sclerosis (Ref. Reference Sumowski117). Thus, although the existence of a genetic component influencing cognition is evident, its relevance in the health and disease processes remains unclear. However, it cannot be denied that its knowledge can bring new insights.
Either way, the investigation of genetic variants affecting cognition and brain structure in healthy people with and without AD neuropathology could be a starting point to determine the intrinsic genetic resistance to dementia. The information obtained through these studies must be comprehensively translated to evaluate its clinical utility in the preclinical stages of AD.
Conclusion
There exists an increasing interest in the characterization of the stages of pre-dementia. Taking into account the high genetic component of AD (Ref. Reference Avramopoulos14), the identification of genetic variants influencing MCI and SCD can provide a new perspective in tackling the disease.
Recent technological improvements have promoted the identification of 28 genetic variants for LOAD. Despite that, data concerning pre-dementia stages remain scarce. At present, APOE gene is the most consistent association with risk to MCI and progression from MCI to AD (Ref. Reference Elias-Sonnenschein54). The CLU locus has also showed promising results (Ref. Reference Lacour60). There are more inconsistent data for SCD. To the best of our knowledge, we are the first to show meta-analysed data evidencing the role of APOE as a risk factor for SCD (Fig. 2).
There is highly pronounced absence of genetic data for pre-dementia stage (Fig. 1). In addition, there is a high degree of heterogeneity between available studies in pre-dementia stage. In an extended way, the MCI studies show a statistical correlation between the genotype and neuropsychological test scores, which mainly provides informative data. In most cases, studies with SCD and MCI individuals have small sample sizes. Moreover, the consideration of population characteristics seems pertinent. SCD and MCI individuals comprise highly heterogeneous population, where converters to AD dementia coexist with converters to other forms of dementia and non-converters. In that scenario, the identification of novel and expected LOAD loci may be hampered by the effect size of the true AD group. That could explain the reduction in the effect size of APOE along stages. On the other hand, the identification of progression factors that are not previously reported must be considered with prudence until their validation. This limitation points to the necessity of using larger cohorts in studies involving a pre-dementia stage.
Efforts are required to provide useful data, which can help in designing strategies to stop or modulate the course of the disease. In that sense, a GWAS in the MCI population seems mandatory. Moreover, the identification of the genetic factors conferring resilience to dementia in non-demented people could provide a good opportunity in uncovering the compensatory mechanisms that may prevent the disease progression. Therefore, a genome-wide approach for endophenotypes involved in CR also seems affordable and advisable.
In conclusion, genetic research in the pre-dementia stages of non-demented people must be potentiated to obtain advances in AD and design prevention strategies.
Acknowledgements
We are indebted to Trinitat Port-Carbó and her family for their support to the Fundació ACE research programs. Fundació ACE collaborates with the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED, Spain) and is one of the participating centers of the Dementia Genetics Spanish Consortium (DEGESCO). The present work has been performed as part of the Biochemistry, Molecular Biology and Biomedicine doctoral program of S. Moreno-Grau at Universitat Autònoma de Barcelona (Barcelona, Spain).
Financial support
This work is supported by Grant PI13/02434 (Acción Estratégica en Salud. Instituto de Salud Carlos III (ISCIII). Ministerio de Economía y Competitividad, Spain), and Obra Social ‘La Caixa’ (Barcelona, Spain).
Conflict of interest
None.




