Introduction
Translation control, a tightly regulated process, plays a critical role in cell growth, proliferation and differentiation. Among the four consecutive stages of translation (initiation, elongation, termination and ribosome recycling), more attention was paid to initiation (Ref. Reference Sonenberg and Hinnebusch1). At this stage, the 43S preinitiation ribosome complex is recruited to the 5′ terminus of mRNA through translation initiation complex eIF4F (Ref. Reference Fischer2). eIF4F is a heterotrimer complex binding the 5′-terminal cap structure 7-MeGpppN (N is any nucleotide). eIF4F consists of eIF4E, the 5′ cap mRNA-binding protein; eIF4A, an ATP-dependent helicase unwinding the secondary structure of mRNA; and eIF4G, a scaffolding molecule serving a docking function in the assembly of eIF4F complex (Ref. Reference Hsieh and Ruggero3). eIF4E mediates the association of eIF4F with cap structure and promotes recruitment of ribosome to the 5′ end of mRNA, playing a vital role in regulating global translation rates. The role of eIF4E in translation regulation unrelated to initiation, such as export of some specific mRNAs (e.g. cyclin D) from nucleus to cytoplasm (Refs Reference von der Haar, Gross, Wagner and McCarthy4, Reference Topisirovic5), has also been emphasised.
Cellular mRNAs can be categorised into two groups according to their structure property and inherent ‘translatability’: strong mRNAs (e.g. housekeeping genes), which have relatively short, unstructured 5′ UTRs (less C + G content); and weak mRNAs, which have lengthy, highly structured 5′ UTRs (G + C rich) (Refs Reference Jia, Polunovsky, Bitterman and Wagner6, Reference Bitterman and Polunovsky7). The significant difference between them is that weak mRNAs are much more dependent on eIF4E availability and poorly translated under normal conditions when eIF4F complex formation is limited. These mRNAs predominantly encode proteins including proto-oncogenes that regulate hallmark capabilities of cancer cells. When eIF4E is overexpressed or hyperactive, translation of weak mRNAs is selectively and disproportionately enhanced, while strong mRNAs are only minimally affected by alteration in eIF4F complex formation.
Several studies have demonstrated that elevated eIF4E levels preferentially increase mRNA translation involved in all aspects of malignancy, such as proto-oncoproteins (e.g. c-myc, cyclin D1, ODC, survivin), angiogenesis factors (e.g. FGF2 and VEGF) and degradative enzymes (e.g. MMP9) (Ref. Reference De Benedetti and Graff8). The list of mRNAs controlled by eIF4E is ever-increasing. Moreover, increased eIF4E up-regulates the nucleocytoplasmic transport of mRNAs encoding potent growth and survival proteins, such as cyclin D1 (Ref. Reference Rousseau, Kaspar, Rosenwald, Gehrke and Sonenberg9). Therefore, eIF4E level affects transformation, tumourigenesis, metastasis, and drug resistance in both experimental cancer models and human cancer tissues. Indeed, its overexpression is common in multiple cancer types, including malignancies of prostate, breast, head and neck, stomach, colon, lung, skin, oesophagus, bladder, cervix and the hematopoietic system (Refs Reference Mamane10, Reference Konicek, Dumstorf and Graff11). Also, elevated eIF4E levels may serve as a biomarker predicting disease progression, overall survival, or relapse after definitive therapy (Refs Reference Flowers12, Reference Hiller13). On the contrary, knockdown eIF4E by small interfering RNA (siRNA) can suppress oncogenic transformation (Refs Reference Zhou14, Reference Soni15, Reference Dong16).
The activation of eIF4E, which functions as a regulatory hub of many major oncogenic pathways, is a crucial event of the PI3K/AKT/mTOR pathway. Consequently, it has attracted considerable attention as a promising target for anticancer drug discovery in practice (Ref. Reference Bitterman and Polunovsky7). This review provides a comprehensive overview of strategies applicable for developing eIF4E-targeted agents.
eIF4E regulation and targeting strategies
eIF4E is regulated at multiple levels, including gene expression, sequestration and phosphorylation, etc. (Refs Reference Martineau, Azar, Bousquet and Pyronnet17).
At the transcriptional level, Myc is one of the best known transcription factors, which can activate eIF4E gene through two Myc-binding sites (E-boxes) in the eIF4E promoter (Ref. Reference Bitterman and Polunovsky7). At post-transcriptional level, HuR, a transcriptional factor, is responsible for stabilising eIF4E mRNA (Ref. Reference Topisirovic18). Post-translationally, eIF4E can be ubiquitinated primarily at Lys-159 and go through proteasome-dependent degradation (Ref. Reference Murata and Shimotohno19).
The interaction of eIF4E with eIF4 G is indispensible for cap-dependent translation initiation. A group of factors generally known as eIF4E inhibitory proteins modulate the eIF4E–eIF4G interaction (Ref. Reference Richter and Sonenberg20). The most well-studied eIF4E inhibitory proteins are 4EBPs, which sequester free-state eIF4E from eIF4G and block eIF4F complex formation. This sequestration results in the repression of translation of certain mRNAs that normally require high levels of available eIF4E (Ref. Reference Richter and Sonenberg20). Upon nutrients, energy, growth factors and stress stimulation, 4EBPs become phosphorylated at different sites as a consequence of the activation of PI3 K/AKT/mTOR signalling pathways. 4E-BP1 is one of the direct substrates of mTOR Complex 1 (mTORC1). Phosphorylated 4E-BP1 releases eIF4E, which is then free to associate with eIF4G to stimulate translation initiation (Refs Reference Tee and Blenis21, Reference Wullschleger, Loewith and Hall22). Besides 4EBPs, the newly discovered eIF4E inhibitory proteins (e.g. Maskin and Cup) associate with eIF4E only on specific mRNAs through interactions with RNA-binding proteins (Ref. Reference Richter and Sonenberg20).
In addition, eIF4E itself has been shown to be phosphorylated in cancer cells, which is a prerequisite for the activity of eIF4E in cancer cells, whereas dispensable for normal development (Ref. Reference Hay23). Thus, an increased level of phosphorylated eIF4E was found in a broad spectrum of cancer cell lines (Ref. Reference Fan24). Phosphorylation of eIF4E (Ser209) is mediated by the MAP kinase-interacting protein kinases (Mnk1 and Mnk2), which are in turn activated by ERK and p38 MAPK pathways (Refs Reference Hou, Lam, Proud and Wang25, Reference Diab26).
Till now, eIF4E-targeted strategies should include: targeting eIF4E synthesis; targeting eIF4F complex integrity (antagonising eIF4E-to-cap and eIF4E-eIF4 G interaction); sequestration of eIF4E and phosphorylations of eIF4E. A summary of the strategies are shown in Figures 1 and 2. Therapeutic agents derived from these strategies that have been developed are summarised in Figure 3 and Table 1 and will be reviewed in detail.
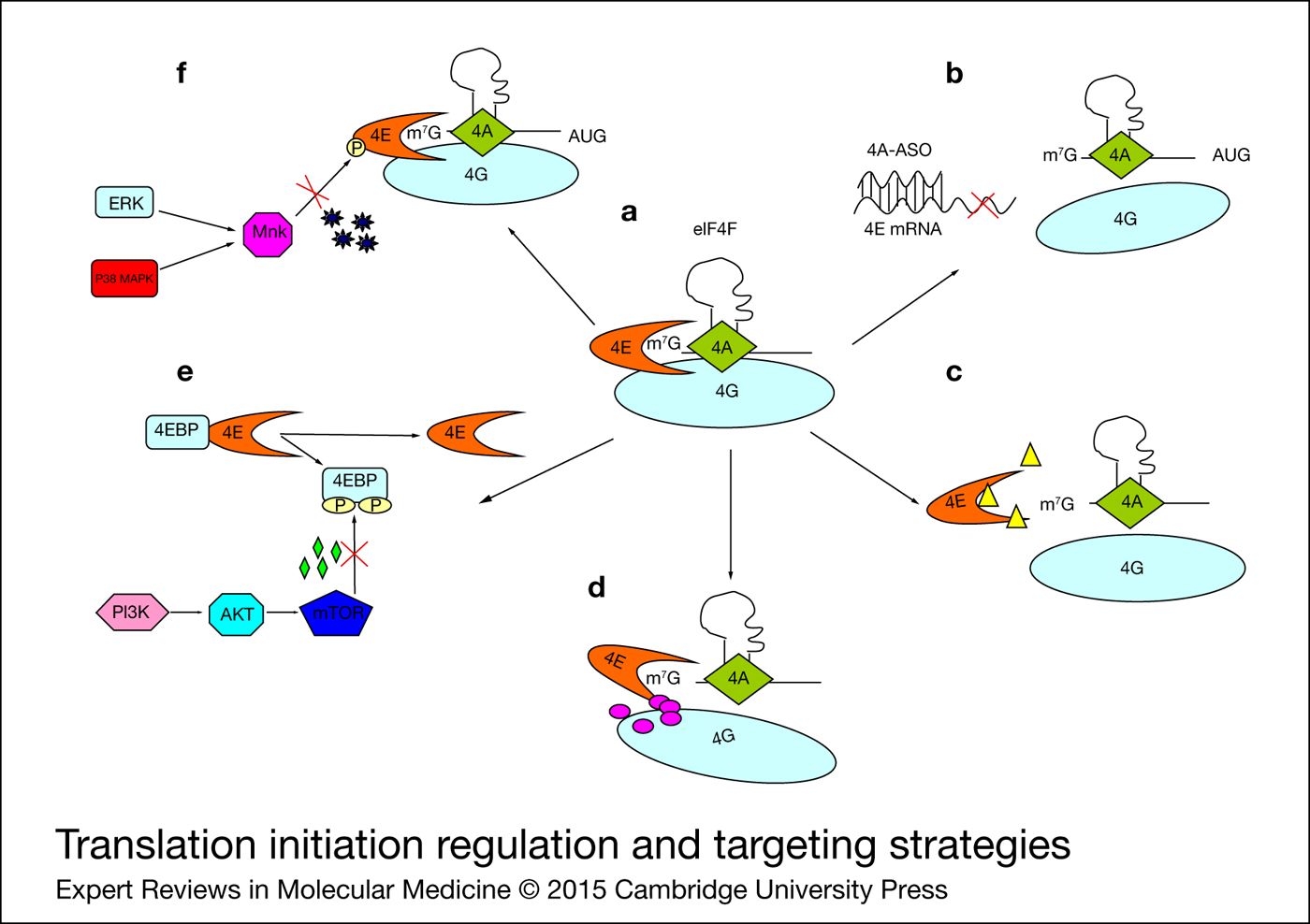
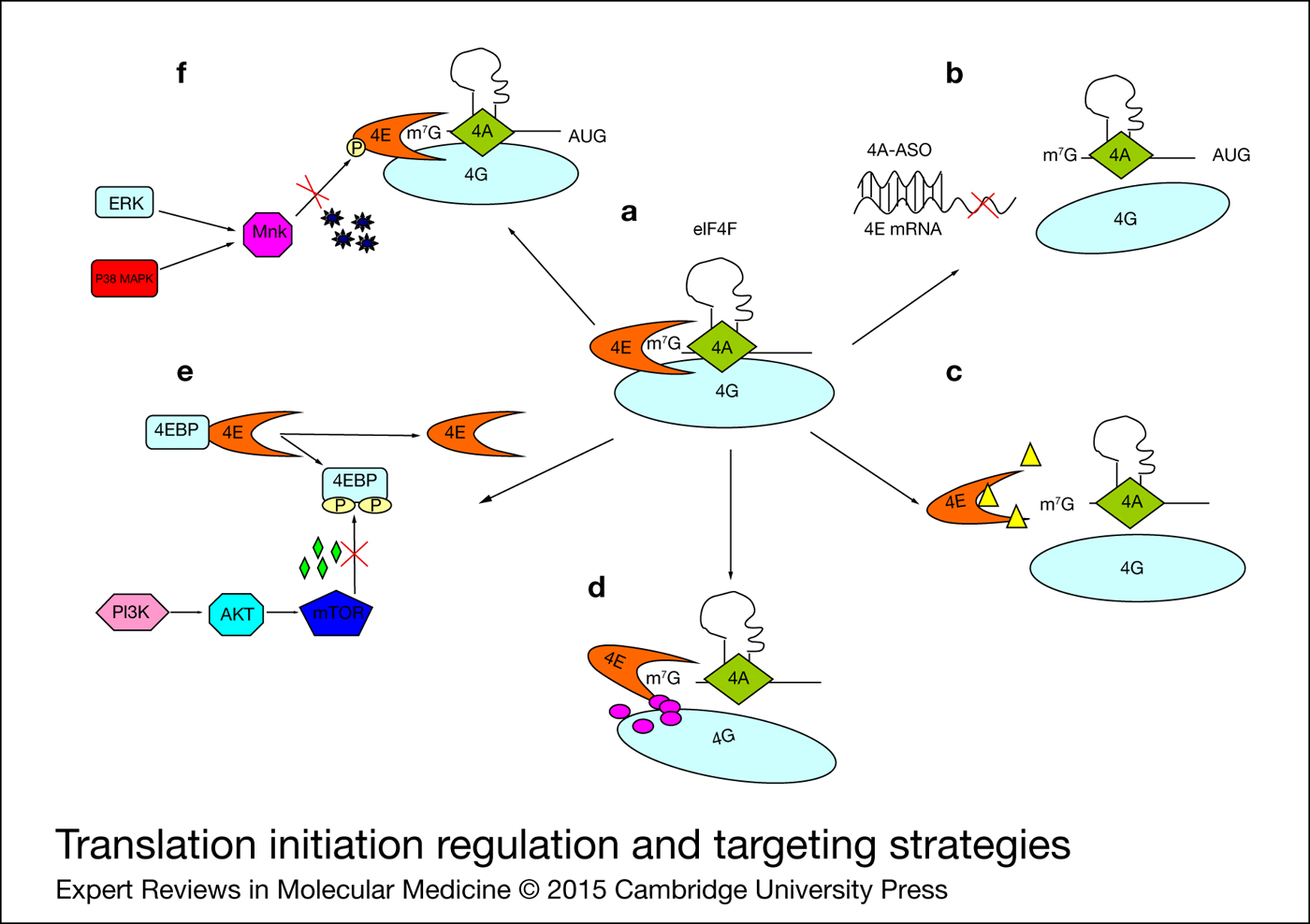
Figure 1. Translation initiation regulation and targeting strategies. (a) Translation initiation complex eIF4F is regulated at distinct levels. (b) siRNA or ASO degrade eIF4E mRNA to reduce its expression. (c) Ribavirin or nucleotide analogues disrupt interactions between eIF4E and Me7G-capped mRNA. (d) Small molecules disrupt eIF4E/eIF4 G association. (e) Memetic peptides or mTOR inhibitors sequester eIF4E. (f) Mnk inhibitors prevent eIF4E activation. 4E, eIF4E; 4G, eIF4G; 4A, eIF4A; 4EBP, eIF4E-binding protein; ASO, antisense oligonucleotide.
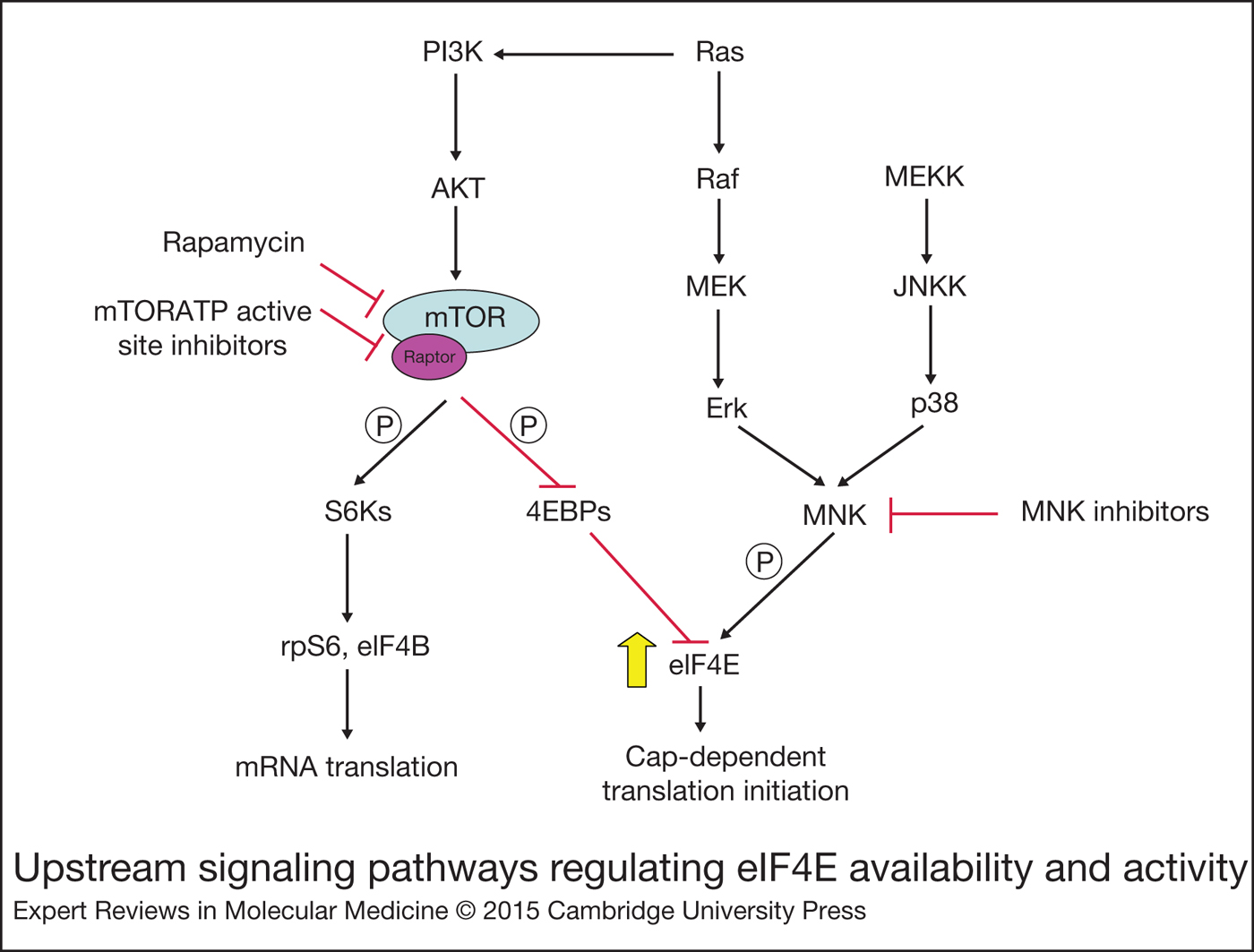
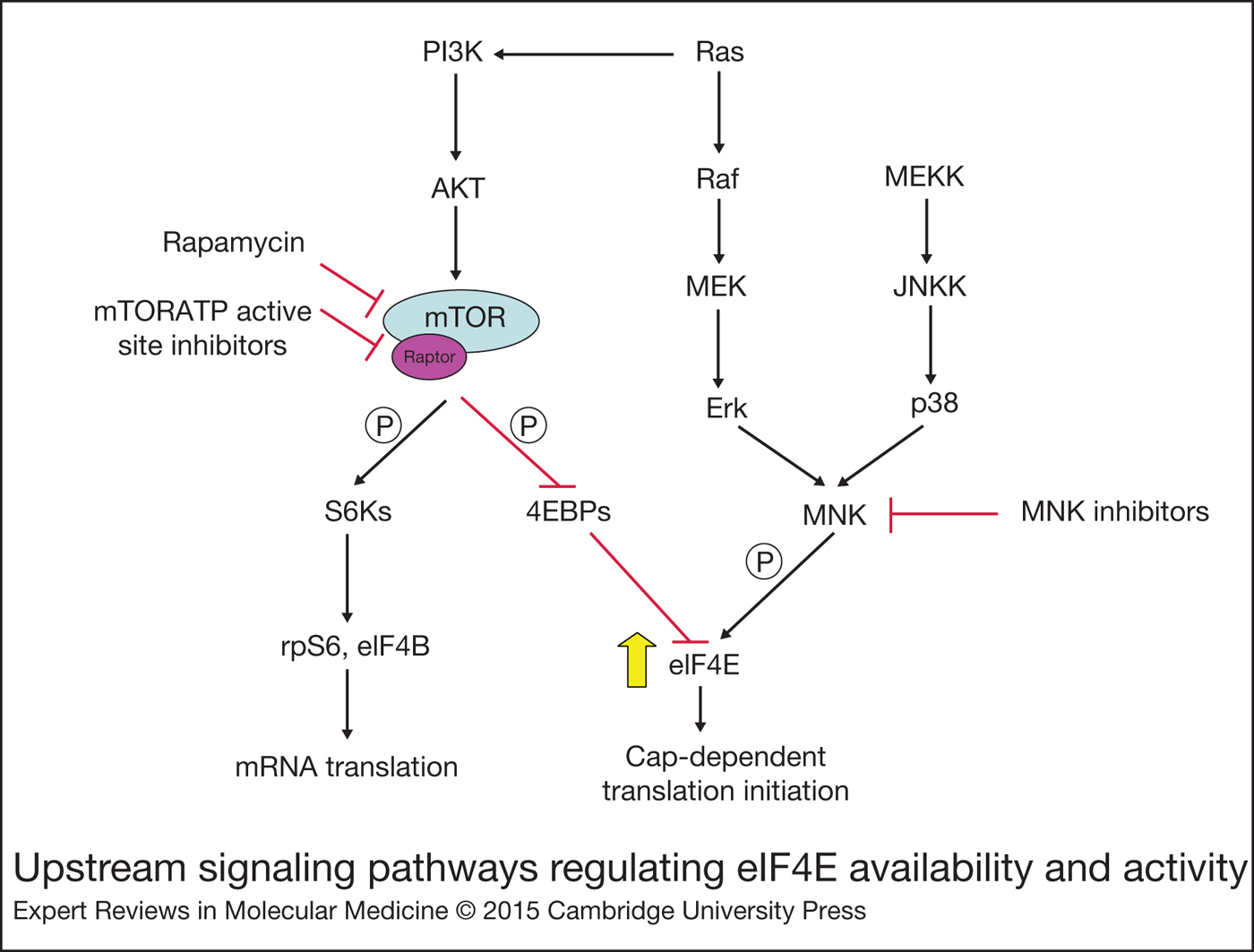
Figure 2. Upstream signalling pathways regulating eIF4E availability and activity.

Figure 3. Chemical structures of eIF4F-targeted agents.
Table 1. Agents targeting eIF4F at different levels

ASO, antisense oligonucleotide; 4EBP, eIF4E-binding protein.
Targeting eIF4E synthesis: siRNA and antisense oligonucleotide (ASO)
eIF4E overexpression leads to the development of lymphomas and other cancers in vitro and in vivo (Ref. Reference Ruggero99), directly targeting eIF4E mRNA by siRNA (Refs Reference Zhou14, Reference Soni15, Reference Choi100, Reference Oridate, Kim, Xu and Lotan101) or specific oligonucleotide (ASO) (Refs Reference Graff27, Reference Hong28, Reference Graff102, Reference Jacobson103) has been extensively studied.
eIF4E knockdown by siRNA resulted in cell cycle arrest, suppression of colony formation, inhibition of cell mobility and enhanced chemosensitivity in MDA-MB-231 triple negative (TN) breast cancer cells (Ref. Reference Zhou14). Although eIF4E knockdown inhibited growth in various breast cancer cell lines, it did not lead to activation of Akt (Ref. Reference Soni15). In addition, eIF4E knockdown can suppress cell growth in endometrial adenocarcinoma and squamous carcinoma (Refs Reference Choi100, Reference Oridate, Kim, Xu and Lotan101). Tumour-specific RNAi via using survivin promoter-driven RNA interference system was recently demonstrated to reduce eIF4E gene expression effectively and specifically, resulting in apoptosis, growth inhibition and enhancement of chemosensitivity to cisplatin in breast cancer cells both in vitro and in vivo (Ref. Reference Dong16). Moreover, eIF4E silencing enhanced radiosensitivity of tumour cells, while has no effect on normal cells (Ref. Reference Hayman104).
Antisense RNA (asRNA), a single-stranded RNA, can be introduced into cells to inhibit the translation of a complementary mRNA by base pairing to it and physically obstructing translation machinery (Ref. Reference Weiss, Davidkova and Zhou105). Down-regulation of eIF4E via asRNA slowed down soft agar growth, increased tumour latency, and accelerated the rates of tumour-doubling times (Ref. Reference Rinker-Schaeffer, Graff, De Benedetti, Zimmer and Rhoads106). asRNA therapy for eIF4E can also be used as adjuvant therapy for head and neck cancers, particularly in cases in which elevated eIF4E is found in the surgical margins (Ref. Reference DeFatta, Nathan and De Benedetti107).
ASOs are unmodified or chemically modified single- stranded DNA molecules. In general, they are relatively short (12–25 nucleotides) and hybridise to complementary target mRNAs by Watson–Crick base pairing (Ref. Reference Dias and Stein108). ASOs have been used to selectively inhibit thousands of genes in mammalian cells and multiple genes in humans. There are over 20 antisense drugs currently in clinical trials, some of which are showing encouraging results (Ref. Reference Dean and Bennett109). First generation ASOs contained a phosphorothioate (a sulfur substitution of a non-bridging O) backbone, whereas second generation ASOs, contained the phosphorothioate backbone plus 2′-O-methoxyethyl modification of riboses at the 5′ and 3′ ends. These modifications enhance affinity for targeted RNA, thus increasing stability and potency, improving antitumour potential and decreasing toxicity (Ref. Reference Mani110). Similarly, eIF4E ASOs were designed specifically to recruit endogenous RNase H and decreased eIF4E expression at the mRNA level (Refs Reference Zhang111, Reference Yu112).
Graff and colleagues designed ASOs capable of targeting both murine and human eIF4E and evaluated their effects on eIF4E reduction in both human xenografts and normal mouse tissues (Ref. Reference Graff27). These ASOs decreased eIF4E proteins and had dramatic cytotoxic effects at nanomolar concentrations across a panel of human cancer cell lines, including prostate cancer, breast cancer, head and neck cancer, non-small cell lung cancer and mesothelioma (Refs Reference Graff27, Reference Graff102, Reference Jacobson103). These ASOs also repressed expression of eIF4E-regulated proteins, inducing apoptosis as well as preventing angiogenesis (Refs Reference Graff27, Reference Graff102, Reference Jacobson103). Most importantly, intravenous ASO administration selectively and remarkably reduced eIF4E expression in human tumour xenografts, significantly suppressing tumour growth without obvious changes of body and organ weight, or liver transaminase levels (Ref. Reference Graff27). A phase I dose escalation design was used to determine the dose level of eIF4E ASO LY2275796 that could be safely administered to patients with advanced solid tumours. LY2275796 was well tolerated at the 1000 mg dose level with only mild toxicities (grades 1–2), which meant it has alluring prospect of clinical application (Ref. Reference Hong28).
Antagonising eIF4E/cap interaction
Ribavirin
Targeting the interaction of eIF4E and 7-MeG-Capped mRNA becomes attractive because an effective compound based on this target should inhibit eIF4E binding to capped mRNA specifically and block translation initiation subsequently. One typical approach to this is ribavirin (1-β-D-ribofuranosyl-1, 2, 4-triazole-3-caboxamide), which shares similar structure to 7-MeGTP (Refs Reference Hofmann, Herrmann, Sarrazin and Zeuzem29, Reference Picardi113). It was observed that ribavirin bound to eIF4E with micromolar affinity and competed with eIF4E:mRNA binding (Ref. Reference Kentsis, Topisirovic, Culjkovic, Shao and Borden30). At the same time, it impaired eIF4E-dependent Akt survival pathways and potently inhibited the biological effect of eIF4E (Ref. Reference Tan, Culjkovic, Amri and Borden31). As a result, it suppressed eIF4E-mediated oncogenic transformation as well as tumour growth both in vitro and in vivo (Ref. Reference Kentsis, Topisirovic, Culjkovic, Shao and Borden30).
The clinical efficacy of ribavirin in M4/M5 acute myeloid leukaemia (AML) patients was promising (Ref. Reference Assouline32) because it can cooperate with a wide variety of established agents to reduce the colony formation in primary AML specimens (Ref. Reference Kraljacic, Arguello, Amri, Cormack and Borden33). In a Phase II trial, ribavirin treatment benefits poor prognosis AML patients (Ref. Reference Borden and Culjkovic-Kraljacic34). 10 mM ribavirin was not cytotoxic to primary chronic lymphocytic leukaemia (CLL) lymphocytes in vitro, and significantly sensitised 76% of the samples tested with fludarabine (Ref. Reference Martinez-Marignac35). Ribavirin also has effects on solid tumour and inhibits growth of several breast cancer cell lines with the elevated eIF4E level (Ref. Reference Pettersson36). More interestingly, ribavirin effectively restored oestrogen receptor alpha (ESR1) gene expression alone and even more in combination with suberoylanilide hydroxamic acid, leading to tamoxifen sensitivity restoration in ESR1 negative breast cancer cell lines (Ref. Reference Sappok and Mahlknecht37). Pharmacologic inhibition of eIF4E with ribavirin also improved tumour cell radiosensitivity (Ref. Reference Hayman104).
However, parallel studies have shown different outcomes. Yan and co-workers found that ribavirin was unable to function as a cap analogue in chemical cross-linking assays, cap-affinity chromatography and cap-dependent translation assays (Ref. Reference Yan, Svitkin, Lee, Bisaillon and Pelletier114). Independently, Westman et al. verified the findings of Yan et al. in a series of experiements (Ref. Reference Westman115). In contrast, Kentsis et al. have rebutted their findings by suggesting specific binding of ribavirin to eIF4E through mass spectrometry detection (Ref. Reference Kentsis116). These contrasting results may likely be because of different experimental methods and conditions.
Other cap-binding antagonists
Over the years, a large variety of nucleoside and nucleotide analogues derived from 7-MeGTP have been synthesised and evaluated for their ability of inhibiting eIF4E binding to capped mRNA specifically. Three independent groups (Refs Reference Cai38, Reference Grudzien117, Reference Chen118) have developed libraries of 7-MeGMP analogues with favourable drug-like properties. Since aryl substitution at N7 has displayed a superior binding affinity (Ref. Reference Cai38), studies have therefore been focused on the utility of the synthetic nucleotide derivative 7-benzyl guanosine monophosphate (7-BnGMP) to block the binding of eIF4E to mRNA cap (Ref. Reference Jia39). A recent crystallographic study with co-crystals of 7-BnGMP and eIF4E revealed that cap-binding pocket undergoes a unique structural change in order to accommodate the benzyl group (Ref. Reference Brown, McNae, Fischer and Walkinshaw40).
While effective in mammalian cell-free systems and zebrafish embryos, the efficacy of 7-BnGMP in cells is poor because of its low cell-membrane permeability. One approach to improve its in vivo activity is to develop a stable, cell-permeable prodrug (pro-nucleotide) which can be bio-activated within cells (Ref. Reference Wagner, Iyer and McIntee119). Phosphoramidates are promising prodrugs for this purpose in consideration of their low toxicity, high solubility and stability. In fact, there have been several successful examples of their applications for antiviral and anticancer therapies (Refs Reference Rawal120, Reference Vernachio121).
Carston R. Wagner's group recently reported the synthesis of a novel class of Histidine Triad Nucleotide Binding Protein (HINT)-dependent pro-nucleotides that interdict epithelial-to-mesenchymal transition (EMT) (Ref. Reference Ghosh41). 4Ei-1, one of the novel prodrugs, powerfully inhibited cap-dependent translation in zebrafish embryos without causing developmental abnormalities, and prevented eIF4E from triggering EMT in zebrafish ectoderm explants without obvious toxicity (Ref. Reference Ghosh41). Metabolism studies with whole cell lysates demonstrated that this prodrug was rapidly converted into active metabolite 7-BnGMP (Ref. Reference Ghosh41). 4Ei-1 is the first nontoxic small molecule able to repress EMT by targeting the interaction of eIF4E with mRNA cap. More recently, Chen et al. pointed out that 4Ei-1 was a novel prodrug that reduced proliferation, suppressed colony formation, diminished association of eIF4E with the mRNA cap, and sensitised mesothelioma cells to pemetrexed (Ref. Reference Chen42). Shui and co-workers showed that treatment of breast and lung cancer cells with 4Ei-1 resulted in chemosensitisation to gemcitabine and induced eIF4E proteasomal degradation, providing another mechanism of 4Ei-1 to induce translation inhibition except for down-regulation of eIF4E cap binding (Ref. Reference Li43).
Targeting eIF4E and eIF4G interaction
Studies have demonstrated that, eIF4E function is also regulated at the level of interactions with eIF4G and 4E-BPs (Ref. Reference Richter and Sonenberg20). This occurs on the dorsal surface of eIF4E, opposite to the cap-binding site. Binding of eIF4 G to eIF4E improves cap-dependent translation through recruitment of eIF4A and the eIF3-40S ribosomal subunit (Refs Reference Martineau, Azar, Bousquet and Pyronnet17, Reference Richter and Sonenberg20). Thus, targeting the eIF4E/eIF4 G protein-protein interaction is a rational mechanism to repress cap-dependent translation.
4EGI-1
The formation of eIF4E/eIF4 G complex is regulated by the 4E-BPs, which competes with eIF4G for binding to eIF4E (Ref. Reference Marcotrigiano, Gingras, Sonenberg and Burley122). Gerhard et al. developed a high-throughput fluorescence polarisation assay for identifying small-molecule inhibitors of eIF4E/eIF4 G interaction to pharmacologically mimic anti-eIF4F effect of 4E-BPs (Ref. Reference Moerke44). Among the 16 000 compounds screened, the most potent one identified is 4EGI-1, which bound eIF4E, disrupted eIF4E/eIF4 G association, and inhibited cap-dependent translation. Interestingly, while 4EGI-1 displaced eIF4 G from eIF4E, it effectively enhanced 4EBP1 association both in vitro and in cells (Ref. Reference Moerke44). 4EGI-1 exhibits pro-apoptotic activity and represses the growth of multiple cancer cell lines. Its treatment caused cell death in Jurkat cell lines and multiple myeloma cells, decreased proliferation of A549 lung cancer cell lines (Refs Reference Moerke44, Reference Descamps45). In primary AML cells, 4EGI-1 dramatically slowed down clonogenic growth of AML precursors and induced apoptosis in massive blast cells (Ref. Reference Tamburini46), which thus represented an attractive option for the development of new therapies in AML. More recently, some researchers attempted to improve the physicochemical properties of 4EGI-1 to meet the urgent demand for clinical application (Refs Reference Takrouri123, Reference Yefidoff-Freedman124). Subsequently, a series of rigidified mimetic of 4EGI-1 were synthesised and characterised, which were more potent than the parent inhibitor (Ref. Reference Mahalingam125).
Although 4EGI-1 was discovered as a small molecule inhibitor that disturbed the interaction of eIF4E and eIF4G, Fan et al. revealed that 4EGI-1 sensitised human lung cancer cells by promoting TRAIL-mediated apoptosis (Refs Reference Fan, Li, Yue, Khuri and Sun126, Reference Panner, Parsa and Pieper127). In addition, cancer cells showed only 2-fold higher susceptibility to 4EGI-1 than their non-transformed counterparts. This inefficient therapeutic index, together with the significant ‘off target’ mechanism, dampens the original enthusiasm for this agent.
4E1RCat
Another small molecule inhibitor 4E1RCat has been discovered (Ref. Reference Cencic47) after screening a library of 217 341 compounds via a time-resolved (TR)-fluorescence resonance energy transfer (FRET) method. 4E1RCat interfered with the interactions between eIF4E and eIF4 G or 4E-BP1. As a consequence, cap-dependent translation is suppressed. This compound can reverse tumour chemoresistance (doxorubicin) in Eμ−Myc lymphoma mouse model by sensitising cells to the proapoptotic action of DNA damage (Ref. Reference Cencic47).
Other agents targeting eIF4E and eIF4 G Interaction
Cao et al. recently reported that Quabain, a kind of cardiac glycoside, directly bound eIF4E, destroyed eIF4E/eIF4 G association, thus inhibiting cap-dependent translation and down-regulating its critical target HIF-1. The association between Ouabain and eIF4E gave us a new clue of using cardiac glycosides for cancer therapeutics (Ref. Reference Cao128). Perillyl alcohol, a kind of secondary product of plant mevalonate metabolism, also attenuated interactions between eIF4E and eIF4 G in prostate cancer cell lines (Ref. Reference Peffley, Sharma, Hentosh and Buechler129).
Sequestering eIF4E
4EBP mimetic peptides directly binding eIF4E
As mentioned above, 4EBP sequesters eIF4E and consequently prevents cap-dependent translation initiation. Reasonably, 4EBP mimetic peptides which directly bind with eIF4E can reduce free eIF4E level, just as 4EBPs do.
Recently, a strategy developed by Naora and co-workers is to use 4EBP-based peptides to sequester eIF4E (Ref. Reference Ko, Guo, Barengo and Naora48). They designed a peptide containing residues 49–68 of 4EBP1, and fused it to an analogue of gonadotropin-releasing hormone (GnRH). GnRH agonist-4EBP fusion peptide efficiently repressed the growth of GnRH receptor-expressing tumour cells, but not receptor negative cells, which is suitable for a targeted strategy. Therefore, GnRH–4EBP fusion peptide has the potential to treat ovarian cancer because this kind of cancer is hardly cured by conventional chemotherapies.
In addition, a eukaryotic expression vector pSecX–t4EBP1 was skilfully constructed, which contained phosphorylation defective 4E-BP1 domain and the protein transduction domain. The former domain down-regulated the expression of eIF4E by direct binding, and the latter domain can help plasmid penetrate the cellular membrane and enhance the efficiency of vector's spread. Interestingly enough, this plasmid significantly down-regulated tumour growth and improved the radiosensitivity of mouse breast carcinoma allografts in BALB/C mice model (Ref. Reference Yang49). Moreover, Terence et al. designed a series of peptides based on the conserved eIF4E-binding motifs linked to the penetratin peptide-carrier sequence. These peptides can effectively bind recombinant human eIF4E in vitro and induce apoptosis after introduction into MRC5 cells (Ref. Reference Herbert, Fahraeus, Prescott, Lane and Proud50).
Rapamycin and Rapalogues
In general, normalising deregulated eIF4F-mediated translation can be accomplished indirectly by interrupting upstream signals leading to eIF4E dissociation from 4EBPs. Naturally, targeting mTOR signalling pathway is a therapeutically attractive option for the purpose of sequestering eIF4E because 4EBPs are direct substrates of mTOR kinase (Ref. Reference Moschetta, Reale, Marasco, Vacca and Carratu130).
So far, rapamycin (sirolimus) and its analogues are the most well characterised mTOR inhibitors. As an immunosuppresive drug, rapamycin can also act as a cytostatic agent, preventing growth of various tumour cell lines including renal cancer, small cell lung cancer, and pancreatic cancer cells (Refs Reference Luan51, Reference Seufferlein and Rozengurt52, Reference Grewe, Gansauge, Schmid, Adler and Seufferlein53). One potential mechanism explaining its antiproliferative effects is the prevention of phosphorylation of 4E-BP1 by mTORC1, which in turn sequesters eIF4E and restrains the initiation of cap-dependent translation (Ref. Reference Guertin and Sabatini54).
Although rapamycin has shown promising antitumour effects in several experimental tumour models, its clinical trials as an anticancer drug is unsuccessful at present. Therefore, several rapamycin analogues (rapalogues) with more favourable pharmaceutical characteristics have been developed, such as CCI-779 (Temsirolimus), RAD001 (Everolimus), AP23573 (Deforolimus,), SAR943 (32-deoxorapamycin) (Ref. Reference Eynott131) and ABT-578 (zotarolimus) (Ref. Reference Karyekar132). CCI-779, RAD001 and AP23573, which are currently under clinical trials, have shown obvious antitumour effects against a diverse range of cancer types in preclinical studies. CCI-779 inhibited mTOR function in several breast cancer cell lines (Ref. Reference Yu58). Antiangiogenic effects may substantially contribute to its antitumour activity in breast cancer (Ref. Reference Del Bufalo59). Also, CCI-779 can potentially be used as an adjuvant therapy in head and neck squamous cell cancer (Ref. Reference Nathan60), which has showed remarkable efficacy in PTEN-deficient tumours (Ref. Reference Neshat61). As such, CCI-779 recently became the first FDA-approved mTOR-targeted agent based on a phase III trial in advanced renal cell carcinoma patients (Ref. Reference Hudes62). In haematologic malignancies, CCI-779 has substantial single-agent activity in relapsed mantle cell lymphoma (MCL) patients (Ref. Reference Witzig63). RAD001, the only orally active rapamycin derivative, possessed antitumour activity in MCL and diffuse large B-cell lymphoma (Ref. Reference Wanner66). A phase II clinical trial demonstrated that AP23573 was well tolerated and had antitumour activity in patients with heavily relapsed or refractory hematologic malignancies (Ref. Reference Rizzieri67).
Rapamycin and rapalogues can also be used in combination with other chemotherapeutics. Simultaneous administration of rapamycin and EKI-785 (EGFR inhibitor) lead to synergistic antitumour effects in glioblastoma (GBM) multiforme cells (Ref. Reference Rao55). Additionally, rapamycin increased the ability of cisplatin to induce apoptosis in human promyelocytic leukaemia cell line HL-60 and human ovarian cancer cell line SKOV3 (Ref. Reference Shi56). Also, it can effectively restore tamoxifen response in breast cancer cells with aberrant Akt activity (Ref. Reference deGraffenried57). CCI-779 was able to restore cisplatin sensitivity in small cell lung cancer cell lines selected for cisplatin resistance (Ref. Reference Wu64). Combination use of CCI-779 and gemcitabine achieved a significantly better survival in xenograft models of human pancreatic cancer compared with CCI-779 or gemcitabine alone (Ref. Reference Ito65).
Second-generation mTOR inhibitors
Extensive research on mTOR has uncovered a complex network of regulatory loops that impact on mTOR-targeting approaches (Ref. Reference Efeyan and Sabatini133) and may explain the inherent limitations of rapamycin-based strategies. For example, when mTORC1 is inhibited and unable to activate S6K, S6K-mediated feedback loop can lead to an up-regulation of PI3K signalling, and provide pro-survival and proliferative signals through Akt (Ref. Reference O'Reilly134). These loops, at least in some degree, counteract the effects of rapamycin in experimental cancer models and in patients (Ref. Reference Efeyan and Sabatini133). Moreover, because mTORC2 also plays a vital role in tumourigenesis (Ref. Reference Guertin135), the high selectivity of rapamycin for mTORC1 (Refs Reference Zoncu, Efeyan and Sabatini136, Reference Harris and Lawrence137) really triggers a major concern. Thirdly, rapamycin does not inhibit 4EBP phosphorylation by mTOR in some cells. One explanation of this is that it works through sterically blocking mTOR access to substrates, which is inefficient for a small substrate like 4EBP, while very efficient for large ones like S6K1(Refs Reference Hara138, Reference Oshiro139, Reference Kim140). All the drawbacks mentioned above indicate that there exists an urgent need to search for second-generation mTOR inhibitors, which can sequester eIF4E more efficiently.
The second-generation inhibitors, which bind to the catalytic sites of mTOR, inhibit kinase activities of both mTORC1 and mTORC2 (Refs Reference Feldman68, Reference Thoreen69, Reference Chresta70, Reference Yu73, Reference Garcia-Martinez74, Reference Xue75, Reference Yu76). The active-site inhibitors of mTOR, PP242 and PP30, suppressed proliferation of primary cells more potently than rapamycin (Ref. Reference Feldman68). Torin1, another highly potent and selective ATP-competitive mTOR inhibitor, impaired cell growth and proliferation to a far greater extent than rapamycin (Ref. Reference Thoreen69). AZD8055 (Refs Reference Chresta70, Reference Zhao71) and WYE-125132 (Ref. Reference Yu73) are both ATP-competitive mTOR inhibitors and have antitumour activity in vitro and in vivo. Combination use of MEK1/2 inhibitor AZD6244 with AZD8055 enhanced the antitumour efficacy relative to the respective monotherapies in nude mouse xenograft models of human lung adenocarcinoma and colorectal carcinoma (Ref. Reference Holt72). Ku-0063794 is a cell permeable and specific mTOR inhibitor, which inhibited mTOC1 and mTOC2 with an IC50 ~ 10 nM (Ref. Reference Garcia-Martinez74). Palomid 529 reduced not only tumour growth, but also tumour angiogenesis and vascular permeability (Ref. Reference Xue75). WYE-354, a novel pyrazolopyrimidine, displayed robust antitumour activity in PTEN null tumours (Ref. Reference Yu76). All the above inhibitors can effectively minimise the feedback activation of Akt by mTORC2 to avoid offsetting their effects of sequestering eIF4E. More importantly, they more potently inhibit 4EBP phosphorylation compared with rapamycin and rapalogues.
Another class of small molecules related to mTOR kinase inhibitors is the mTOR and PI3 K dual specificity inhibitors. These molecules, such as SF1126 (Ref. Reference Garlich77), NVP-BEZ235 (Refs Reference Maira78, Reference Schnell79), PI103 (Ref. Reference Zou80), XL765 (Ref. Reference Yu76) and GNE-477 (Ref. Reference Heffron81), simultaneously target ATP binding sites of mTORC and PI3 K with similar potency. NVP-BEZ235 and XL765 are undergoing clinical phase I trials. These inhibitors have the unique advantages to avoid PI3 K pathway reactivation caused by mTOR–p70S6 K negative feedback loop, so that they can exert their effects more thoroughly derived from sequestering eIF4E.
Targeting eIF4E phosphorylation
Role of Mnks in tumourigenesis and its regulation
Although some success has been achieved on the inhibition of PI3 K/mTOR axis, the multiple feedback loops make this pathway disappointing, to some degree. The ideal goal should be to down-regulate the function of specific pathway in cancer cells without affecting normal cells and eliciting feedback loops that could impair the therapeutic efficacy. Phosphorylation of eIF4E by Mnks on Ser209 is critical for oncogenic activity of eIF4E (Refs Reference Wendel141, Reference Topisirovic, Ruiz-Gutierrez and Borden142) Therefore, eIF4E phosphorylation has been established as a crucial event in tumourigenesis (Refs Reference Diab26, Reference Furic143), such as prostate cancer progression (Ref. Reference Furic143). Reasonably, targeting Mnks could be the attractive therapeutic approach that in mammalian cells, Ser209 phosphorylation is not essential for the activity of eIF4E in normal cells but is required in cancer cells (Ref. Reference Hay23). Takeshi and co-workers confirmed this viewpoint by demonstrating that Mnk1 and Mnk2 double knockout dramatically delayed the tumour development.
Mnk–eIF4 G interaction plays an essential role in the eIF4E phosphorylation regulation. Mnk interacts with the scaffolding protein eIF4G, which also binds eIF4E and brings Mnk and its substrate into physical proximity (Refs Reference Li144). From the aspect of negative regulation, Mnks can be dephosphorylated and deactivated by protein phosphatase 2A (PP2A), preventing further eIF4E phosphorylation (Ref. Reference Li144).
Combination of targeting Mnk–eIF4E and mTOR pathways
Mnk-induced eIF4E phosphorylation is closely related to mTOR pathway inhibition (Refs Reference Shi145, Reference Wang146). Two research groups have proposed a new strategy of enhancing mTOR-targeted cancer therapy through the combined treatment with Mnk inhibitors. Michal Marzec and co-workers reported that simultaneous inhibition of mTORC1 and Mnk markedly induced apoptosis of cutaneous T cell lymphoma cells (Ref. Reference Marzec147). The combined treatment also arrested the cell cycle progression and suppressed the growth of prostate cancer cells (Ref. Reference Bianchini148). Furthermore, co-exposure to MNK1 and mTORC1 inhibitors profoundly decreased 4EBP1 phosphorylation, protein synthesis and proliferation in glioma cells, and reduced tumour growth in an orthotopic GBM mouse model (Ref. Reference Grzmil149).
Mnk inhibitors
So far, three well-studied Mnk inhibitors have been reported: CGP052088, CGP57380 and Cercosporamide.
CGP052088 is a derivative of staurosporine, a broad-spectrum kinase inhibitor. It inhibited Mnk1 with an IC50 value of 70 nM in biochemical assays and was cytotoxic with a GI50 value of 4.5 µM in a 24 h-MTT proliferation assay (Ref. Reference Tschopp82). CGP57380 was also found to be a potent Mnk1 and Mnk2 inhibitor in vitro (Refs Reference Buxade, Parra-Palau and Proud83, Reference Knauf, Tschopp and Gram84). Its antiproliferative effects in five breast cancer cell lines were primarily cytostatic, rather than cytotoxic, and were potentially because of the inhibition of cyclin D1 synthesis (Ref. Reference Wheater, Johnson and Blaydes85). In addition, CGP57380 is therapeutically useful in blast crisis CML (Ref. Reference Lim86). It enhanced imatinib activity against CML and overcame imatinib resistance through impairing polysome assembly (Ref. Reference Zhang87). Similarly, treatment of pancreatic cancer cell lines MiaPaCa2 and PT45P1 with CGP57380 in combination with gemcitabine caused a greater apoptotic cell death when compared with the use of either CGP57380 or gemcitabine alone (Ref. Reference Adesso88). Another Mnk inhibitor is Cercosporamide, an effective antifungal agent and phytotoxin extracted from the fungus Cercosporidium henningsii (Ref. Reference Sussman89), which suppressed AML precursors and enhanced the antileukaemic properties of cytarabine or mTOC1 inhibitors (Ref. Reference Altman90). Furthermore, it can significantly down-regulate outgrowth of experimental B16 melanoma pulmonary metastases and subcutaneous HCT116 colon carcinoma xenograft tumours, without side effect on body weight. These findings confirmed that Mnk inhibition may provide a tractable cancer therapeutic approach (Ref. Reference Konicek91).
More recently, a novel Mnk inhibitor retinamides (RR), one of retinoic acid metabolism blocking agents, blocked eIF4E phosphorylation and subsequently restrained cell growth, colonisation, invasion, and migration, as well as induced apoptosis in TN and Her-2 overexpressing breast cancer cells through degrading Mnks rather than inhibiting its kinase activity just like the three Mnk inhibitors mentioned above (Ref. Reference Ramalingam150). Further, a series of 5-(2-(phenylamino pyrimidin-4-yl) tiazole-2(3H)-one derivatives have been discovered as selective Mnk2 inhibitors. They facilitated apoptosis in MV4-11 AML cells by reducing the expression of an antiapoptotic protein Mcl-1 (Ref. Reference Diab151).
Conclusions and outlook
Our understanding of human cancer as a multi-factor-network disease has led to the development of next-generation therapeutics. It is clear now that targeting regulatory hubs in the cancer signalling network instead of targeting individual genetic alterations will be more effective in treating a very heterogenous tumour. Accumulating evidences indicate that one of such hubs is eIF4E, serving as a node on which multiple oncogenic signalling pathways converge. As a result, eIF4E and translation initiation provide a promising target for cancer therapeutics. Indeed, enthusiasm for developing small molecule inhibitors blocking eIF4E function has lasted over the years.
In addition to the therapeutic strategies targeting eIF4E aforementioned, some other components of eIF4F complex should be considered as appealing oncology drug targets. These include eIF4A, an RNA helicase, which is frequently activated in cancer cells, either by its overexpression or by repression of the tumour suppressor Pdcd4 (programmed cell death 4) (Ref. Reference Jansen, Camalier, Stark and Colburn152). Silvesterol and Pateamine A are both eIF4A inhibitors. Silvesterol can effectively modulate the activity of eIF4A and repress translation initiation, exhibiting powerful anticancer activity in human breast cancer and prostate cancer xenograft models by inducing apoptosis and inhibiting angiogenesis (Refs Reference Cencic92, Reference Kim93). In addition, the suppressive effects of Pateamine A (PatA) on translation are mediated through increasing the RNA-binding affinity of free eIF4A, thus sequestering eIF4A from the 4F complex, which may lead to stalling of initiation complexes (Ref. Reference Bordeleau94).
The binding of initiator tRNA to the 40S ribosomal unit is mediated by translation initiation factor 2 (eIF2). Phosphorylation of α-subunit of eIF2 prevents formation of the eIF2/GTP/Met-tRNA complex and stops global protein synthesis (Ref. Reference Kimball153). As a consequence, eIF2 is also a promising drug target at the level of translation initiation. eIF2α can be phosphorylated by haeme-regulated inhibitor, PERK/PEK, and the double-stranded RNA-activated protein kinase (PKR) (Ref. Reference Jagus, Joshi and Barber154). So far, reported eIF2 inhibitors include eicosapentaenoic acid (EPA) (Ref. Reference Palakurthi95), clotrimazole (Ref. Reference Aktas96), troglitazone (Ref. Reference Aktas97) and flavonoids (e.g. Genistein and Quercetin) (Ref. Reference Ito, Warnken and May98).
Despite the encouraging targeting potential of cap-dependent translation, our understanding of the role of eIF4E and other members regulating translation initiation in tumourigenesis remains rudimentary. For example, eIF4E is responsible for the regulation of multiple mRNAs involved in cancer progression; however, it is unclear if any of these eIF4E targets is indispensable. A comprehensive mapping of eIF4E target mRNAs will be imperative to elucidate the translational signature of eIF4E and its significance in human cancer. On the other hand, in order to improve the anticancer efficacy, future studies are needed to address the combination usage of eIF4E inhibitors with inhibitors targeting other oncogenic signalling pathways.
Acknowledgement
We thank Dr. Chunhong Yan for critical reading of this paper.
Financial Support
This study was supported by the National Natural Science Foundation of China (grant numbers 31372207 and 31572265); the National Supporting Projects for Science and Techniques (grant numbers 2012BAD12B10 and 2012BAD12B1003); and the Anhui Natural Science and Technology Program of Anhui Province (grant numbers 1208085MC41 and KJ2013A310).
Conflict of Interest
None.
Ethical Standards
I assert that all procedures contributing to this work comply with the ethical standards of NIH guidelines (NIH Pub. No. 85–23, revised 1996) and approved by Animal Care and Use Committee of Anhui Normal University. Approval number is ‘#20150131’.